原文内容
反馈意见
复制下面的地址分享给好友
30天免登录
忘记密码?
首页 > 资讯 > 识林回顾 2022年全球法规变化
2023-02-07
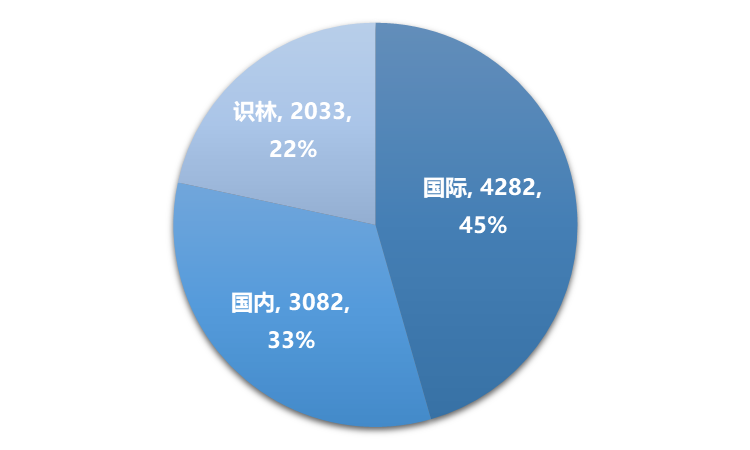
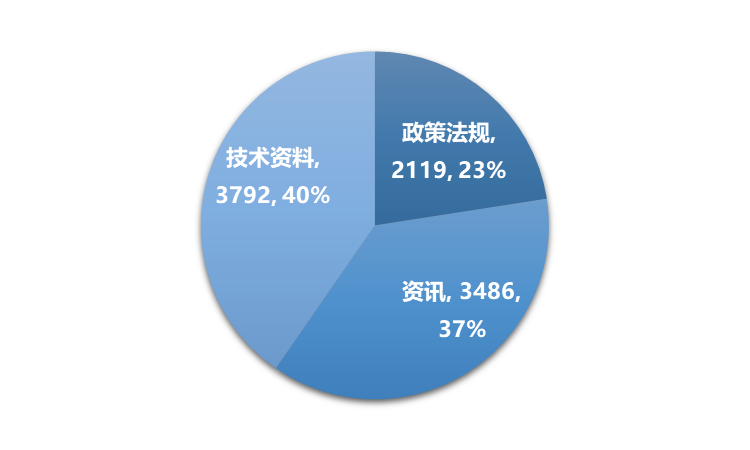
2022年,识林共新增9397篇内容,其中国际新增4282篇,占45%;国内新增3082篇,占33%;识林更新2033篇,占22%。新增内容中,技术资料3792篇,占40%,资讯3486篇,占37%,政策法规及解读2119篇,占23%。
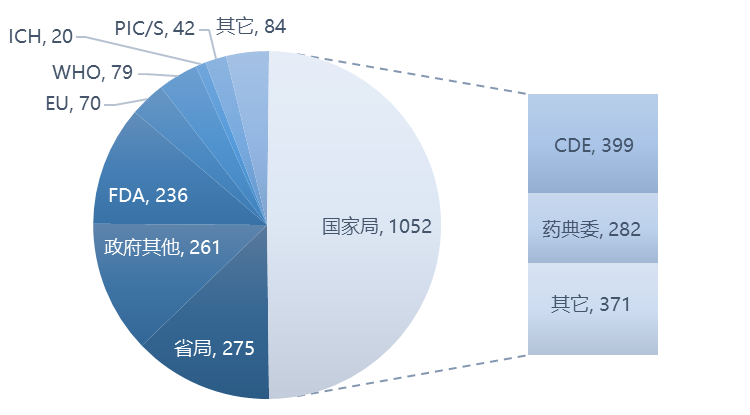
在新增的2119篇政策法规及解读中,国内发布的占75%,包括 WHO、ICH、PIC/S、美国 FDA、欧盟、英国 MHRA、澳大利亚 TGA、日本PMDA等国际机构发布的占25%。
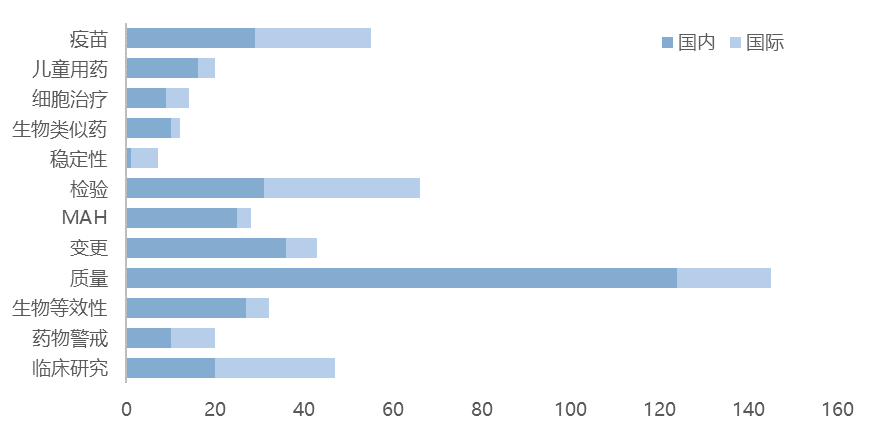
在新增的2119篇政策法规及解读的热点话题来看,质量相关的最多,有将近150篇;受疫情影响,疫苗相关的政策法规及解读,国内外都有将近30篇,儿童用药和细胞及基因治疗是热度较高的产品。国内发布生物类似药相关的指南10篇,远高于国际。MAH的话题国内有超过20篇,也远高于国外。变更话题相关的政策法规及解读也有超过40篇。更多关于2022年新增的政策法规的详细分类,欢迎登录识林的法规指南数据库查找。
对于高度监管的制药行业,法规指南有变,则行业必随之而变。
法规指南之“变”,包括新正式文件落地生效,也包括新征求意见稿发布。前者直接改变许多岗位的每日工作,牵一发动全身,也将改变药品生命周期的方方面面;后者则可能改变思维方式,引发新思考,产生新期待,预示全行业的进步方向。
下面,我们就从国内与国际,正式文件与征求意见这四个维度,简要回顾一下2022年识林用户最关注的政策法规:
2022年,国内监管体系,从顶层设计到具体实施,都在不断进化。年报与召回进一步完善监管闭环;药品注册则将全面进入电子化时代;MAH主体责任落地,给广大Biotech提出了更高要求...最引人瞩目的征求意见稿自然是药管法实施条例,创新药临床领域,除了不断发布的个药原则,“真实世界”、“以患者为中心”、“获益风险评估”等关键概念将影响临床开发的方方面面......
国内法规尚有诸多涉及监管顶层设计的“大变化”,与之相比,国际监管机构更多的是更新各类指南,逐步优化药品生命周期监管体系。2022年定稿的GMP中,热度最高的首推欧盟GMP无菌药品附录;FDA则是从细处着眼,时隔16年修订OOS,此外,FDA修订了批准前检查和药品生产检查的内部规范,志在进入美国市场的国内药企十分关注;论及影响面之广,年度最热莫过于ICH的Q2分析方法验证与Q14分析方法开发,众多分析研发与QC人员都将面临思维与工作的双重变革;从创新角度,FDA年初发布的CAR-T和基因治疗产品指南草案,也许关注人数不如前者,代表的是业界最前沿的监管理念......
识林®版权所有,未经许可不得转载。
法规指南解读:EMA Concept Paper on the revision of Annex 11
适用岗位(必读):
工作建议:
文件适用范围:本文适用于欧盟/欧洲经济区成员国及药品检查合作计划(PIC/S)参与机构的药品GMP附录11计算机化系统的修订。涉及化学药、生物制品等药品类型,包括创新药、仿制药、原料药等注册分类,由EMA和PIC/S发布,适用于Biotech、大型药企、跨国药企等企业类别。
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
本文适用于所有人用药产品,包括化学合成药品和生物制品,重点关注含有化学合成API或生物API的药品。适用于在欧盟区域内的Biotech、大型药企、跨国药企、CRO和CDMO等各类企业。
必读岗位:
文件适用范围:本文适用于无菌药品的生产,包括化学药品、生物制品等,特别关注在欧盟(EU)的生产企业,包括Biotech、大型药企、跨国药企等。
岗位必读建议:
文件适用范围:
本文适用于化学药品和生物制品的新药申请(NDA)、生物制品许可申请(BLA)以及仿制药申请(ANDA),由美国食品药品监督管理局(FDA)发布,主要针对大型药企、Biotech公司、跨国药企以及CRO和CDMO等企业。
文件要点总结:
产品质量评估与风险获益考量:强调FDA在产品开发研究、生产过程和控制策略方面对药品质量的一致性要求,以及在审批过程中对风险和获益的综合评估。
法规与监管要求:明确了根据FD&C法案和PHS法案,药品和生物制品在申请审批前必须满足的安全、有效、纯度和质量的法规要求。
产品质量问题的评估原则:提出了在产品评估中考虑治疗背景、潜在获益和产品质量相关风险的相互关系,以及评估产品质量问题所带来的风险。
未解决质量问题的处理:在某些情况下,即使存在未解决的质量问题,如果产品的获益明显大于风险,FDA可能会批准申请,并允许在批准后提交某些信息。
ANDA的特殊考量:对于ANDA,由于依赖于已确定安全有效的RLD,其产品质量评估可能与NDA和BLA不同,但同样需要满足质量一致性的要求。
岗位职责解读:
文件适用范围:本文适用于化学药品和生物制品的NDA(新药申请)及ANDA(仿制药申请),包括原料药和成品制剂。主要针对美国市场,由FDA发布,适用于大型药企、Biotech公司、跨国药企以及CRO和CDMO等企业。
法规指南解读
适用岗位:
适用范围:
检查流程更新:强调了对批准前检查流程的更新,包括ICH Q10和Q12的整合,亚硝胺杂质控制,以及评估设施的替代工具。
数据报告要求:明确了人用药品行业代码,以及与NDA和ANDA相关的特定检查代码,确保数据的准确报告。
检查结果沟通:规定了检查结果的沟通流程,包括FDA 483表格的回复和EIR的完成。
监管/行政策略:提出了监管建议,包括批准或暂不批准的建议标准。
样品相关报告要求:强调了样品收集和分析的重要性,以及实验室结果的记录和报告。
适用范围:本文适用于化学药、生物制品、放射性药物、原料药等各类药品的生产检查,包括创新药、仿制药、生物类似药等注册分类。适用于美国境内外的药品生产企业,包括Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。发布机构为美国FDA。
文件要点总结:文件强调了对药品生产过程中质量保证的重视,特别是对ICH指南Q9、Q10和Q12的整合,以及对亚硝胺杂质控制的新增要求。明确了药品生产检查(DPI)的类型,包括全面检查和简化检查,并详细描述了不同检查类型的具体操作和要求。特别指出了对药品生产过程中质量风险管理的重要性,以及对药品生命周期管理的技术和监管考虑。文件还提到了替代工具的使用,用于评估设施的合规性。此外,文件详细说明了检查结果的报告要求,包括对重大问题的及时报告和对检查结果的分类处理。最后,文件强调了对药品质量体系的评估,以及对成熟质量实践的观察,这些实践可能超出CGMP要求,显示了一个先进的质量体系。
适用范围:本文适用于化学药品、生物制品和生物类似药的NDA、ANDA和BLA的CMC信息批准后变更。适用于美国FDA监管的药品,包括创新药、仿制药、原料药等。适用于大型药企、Biotech公司、跨国药企以及CRO和CDMO等企业。
在解读FDA发布的《Good ANDA Submission Practices Guidance for Industry》文件时,以下是对该指南的概要解读:
适用范围:本文适用于化学药品的仿制药(ANDA),特别针对美国市场的药物注册分类,适用于Biotech、大型药企、跨国药企等。
法规指南解读:
文件适用范围:本文适用于化学药品的实验室测试,包括原料药、辅料、中间体和成品的测试。适用于由CDER监管的化学药品,不包括生物制品或疫苗。主要针对传统的药品测试和放行方法。发布机构为美国FDA,适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。
适用岗位及工作建议:
文件适用范围:本文适用于化学药和生物制品的创新药、仿制药、生物类似药及原料药,由美国FDA发布,适用于Biotech、大型药企、跨国药企以及CRO和CDMO等企业类别。
文件要点总结:FDA发布的这份指南强调了在药品研发过程中识别和理解患者需求的重要性。文件详细讨论了定性和定量研究方法,以及混合方法研究的设计和实施,以收集患者体验数据。特别强调了在设计研究和问卷时,避免引导性问题,确保问题表述清晰、无歧义,并且要考虑不同患者群体的特殊需求,如儿童、残疾人士和多文化背景的患者。指南还提出了使用社交媒体作为数据收集渠道的考虑因素,包括研究设计的选择、数据收集和分析方法,以及数据质量的评估。此外,指南鼓励与FDA早期互动,以获得关于患者体验数据收集的反馈,帮助企业在药物开发和监管决策中更好地考虑患者的声音。
适用范围:本文适用于包括化学药、生物制品在内的各类药品,特别关注创新药和仿制药的开发。适用于美国FDA监管框架下的Biotech、大型药企、跨国药企以及CRO和CDMO等企业类别。
要点总结:本指南提供了关于选择、开发或修改适合目的的临床结果评估(COA)的指导,以确保在临床试验中准确测量对患者重要的结果。强调了COA的适用性取决于其在特定使用背景下的验证水平,要求申办者提供明确的证据支持COA的适用性。指南提出了一个路线图,指导申办者理解疾病或症状、概念化临床效益和风险,并选择合适的COA类型。特别强调了COA的评分方法应适当,能够反映患者在使用背景下随时间的临床意义变化。此外,指南还讨论了COA的敏感性、解释性和沟通的清晰度,以及如何通过认知访谈和可用性测试来评估COA的理解和负担。最后,指南鼓励申办者在COA开发过程中与FDA进行早期和持续的沟通,以确保COA的适宜性。
适用范围:本文适用于化学药和生物制品,包括创新药、仿制药和生物类似药,以及原料药。适用于在美国进行注册分类的药品,包括Biotech、大型药企、跨国药企以及CRO和CDMO等企业类别。
要点总结:本指南强调了FDA对使用真实世界数据(RWD)和真实世界证据(RWE)以支持药品和生物制品监管决策的当前看法。指南明确了RWD和RWE的定义,RWD是常规收集的与患者健康状况和/或医疗保健提供相关的数据,而RWE是从RWD分析中得出的关于医疗产品使用和潜在获益或风险的临床证据。指南提出了在提交文件中识别RWD/RWE的具体建议,包括使用目的、涉及的研究设计以及RWD的来源类型。特别指出,如果RWD/RWE数据不打算支持产品标签,则不应在提交给FDA的文件中标明。此外,指南还提供了一个附录,作为包含RWD/RWE的提交信息的样本,以帮助申办者和申请人遵循这些指导原则。
文件适用范围:本文适用于化学药和生物制品的生物分析方法验证及样本分析,包括非临床毒代动力学研究、非临床药代动力学研究、临床试验各阶段,以及比较生物利用度/生物等效性研究。适用于ICH地区监管机构。
文件适用范围:本文适用于化学药品和治疗蛋白的原料药与制剂的连续制造(CM),包括新药、仿制药、生物类似药等。适用于采用连续制造技术的企业,包括Biotech、大型药企、跨国药企等。由ICH发布,适用于全球药品监管机构。
文件适用范围:本文适用于化学和生物/生物技术药物的商业化药物物质和产品的放行及稳定性测试中的新或修订的分析程序。同样适用于作为控制策略一部分的其他分析程序,遵循基于风险的方法。科学原理可以在临床开发期间适当地应用。
本文适用于化学药、生物制品、疫苗、中药等药品类型,包括创新药、仿制药、生物类似药、原料药等注册分类。适用于跨国药企、大型药企、Biotech、CRO和CDMO等企业类别。发布机构包括中国、美国、欧盟、印度等。
适用范围:本文适用于生物制品,包括源自人或动物细胞系的生物治疗药物和某些生物制品,涵盖了利用重组DNA技术生产的生物技术产品,如干扰素、单克隆抗体和重组亚单位疫苗。适用于大型药企、Biotech公司、跨国药企以及CRO和CDMO等企业类别。发布机构为国际协调会议(ICH)。
文件要点总结:本文强调了生物技术产品病毒安全性评价的重要性,提出了从原材料选择、生产工艺评估到最终产品病毒检测的全面框架。特别指出了病毒污染的潜在来源,包括原始细胞系和生产过程中意外引入的病毒。明确了对主细胞库(MCB)和工作细胞库(WCB)进行病毒检测的建议,包括逆转录病毒和其他内源性病毒的检测,以及对未处理的散装产品中病毒的检测。提出了病毒清除研究的设计和执行要求,强调了使用不同方法进行病毒灭活或清除的重要性,以达到最大的病毒清除效果。新增了对连续生产过程中病毒安全性的考量,强调了对病毒清除步骤的验证和风险评估的必要性。本文还提供了关于如何使用先验知识来评估病毒清除的指导,以及在生产过程发生显著变化时重新评估病毒清除的要求。
适用岗位必读指南:
文件适用范围:本文适用于所有药品类型,包括化学药、生物制品、疫苗、中药等,适用于创新药、仿制药、生物类似药、原料药等注册分类,由中国国家药监局发布,适用于Biotech、大型药企、跨国药企等各类企业。
文件适用范围:本文适用于人类使用的化学药、生物制品等药品,包括原料药和活性物质。适用于PIC/S成员国的药企,包括Biotech、大型药企、跨国药企等。
根据您提供的文件内容,以下是对WHO_ECSPP_TRS_1044_56th_report_202212的解读:
适用岗位必读:
文件适用范围:本文适用于化学药、生物制品、疫苗和中药等药品类型,特别关注创新药、仿制药、生物类似药和原料药等注册分类。适用对象包括Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。发布机构为世界卫生组织(WHO)。
反馈意见