首页
>
资讯
>
FDA速释制剂BA和BE豁免指南定稿比对解读
出自识林
2018-05-09
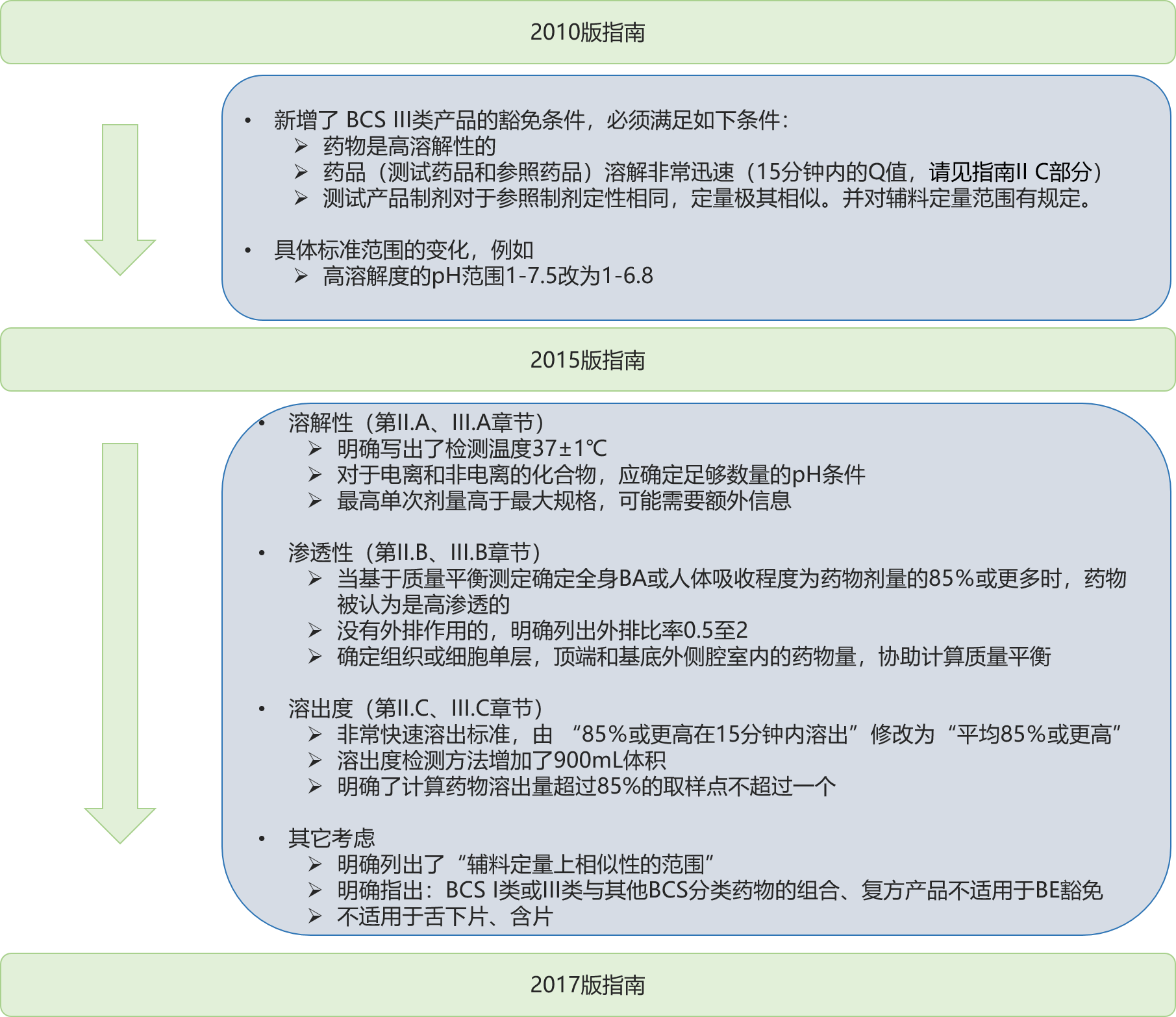
FDA指南,生物药剂学分类中,题为《基于生物药剂学分类速释口服固体制剂的体内生物利用度和生物等效性研究的豁免指南 》(Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System),于2015年5月 发布了修订版指南草案,2017年12月 发布了指南定稿。一图读懂三个版本的变化:
2017版在指南2015年基础上,更多是澄清(clarification)性内容。详见识林(花脸稿 ),导读如下:
溶解性(第II.A、III.A章节)
第II.A章节,溶解性 检测说明了 37±1℃;一直都是这样做的,这里明确写了出来。
第III.A章节,增加一句“A sufficient number of pH conditions should be determined for both ionizable and non-ionizable compounds.”
说明:电离特性的,pH应基于pKa, pKa + 1, pKa - 1, 1和6.8;对于电离和非电离的化合物,应确定足够数量的pH条件。
第III.A章节,第一段最后一句,2015年指南草案仅规定:将药物添加到缓冲液后,应确认溶液的pH值。而2017年指南补充了细节:将药物添加到缓冲液后,应确认溶液的pH值(如果需要,应测量并调整至目标pH值)。另外在平衡溶解度研究结束时,还应测量溶液pH值。
第III.A章节,增加一段“对于服用最高单次剂量高于最大规格的,可能需要额外的信息。如果溶解度分类可能以最高单次剂量作为标准而改变,则需要覆盖治疗剂量范围的宽剂量范围内的额外PK剂量比例信息。”因为EMA Investigation of bioequivalence 指南对生物等效性试验、高溶解度的判断是基于最高单次剂量,可能FDA是希望注意这一点。
渗透性(第II.B、III.B章节)
第II.A章节,增加了“the systemic BA or”,2017年指南指出:当基于质量平衡测定确定全身BA或人体吸收程度为药物剂量的85%或更多时,药物被认为是高渗透的。
第III.B.2. 章节“肠道渗透性 检测方法”,外排转运系统增加了BCRP, MRP2;对没有外排作用的,明确列出即:外排比率0.5至2(i.e., efflux ratio 0.5 to 2)。
第III.B.2.章节,由2015年的linear (pharmacokinetic) relationship,改为2017年的 proportional relationship 和 linear PK of a drug。
第III.B.2. 章节,2017年指南新增:在体外测试结束时,应确定组织或细胞单层,顶端和基底外侧腔室内的药物量,以协助计算质量平衡。如果从顶端和基底外侧腔室的复原 >80%,则没有必要测定组织或细胞单层中的药物。
溶出度(第II.C、III.C章节)
第II.C章节,对于非常快速溶出的标准,由 “85%或更高在15分钟内溶出”修改为“平均85%或更高”。
第III.C章节“判断溶出度 特征”,溶出度检测方法增加了900mL体积,但还是500mL常用。
溶出度检测仪器应符合的要求,除了USP 条款外,新增“FDA's guidance on Mechanical Calibration of Dissolution Apparatus 1 and 2”。
增加了一句:计算药物溶出量超过85%的取样点不超过一个。Only one measurement should be considered after 85 percent dissolution of both products。这个要求是企业一直都是这么做的。
多处增加了对拟定的每个规格(for each strength)都要做试验,以支持产品是快速、非常快速的溶出;以支持biowaiver。对此一直都是这么做的。
其它考虑
第V.A章节,BCS 3类豁免的第3个条件,指南草案写的是:“用量必须在SUPAC IR指南 对于1级或2级变更允许的范围之内”; 2017年的指南定稿第V.A.章节明确列出了“辅料定量上相似性的范围”。
第V.C章节明确指出:BCS I类或III类与其他BCS分类药物的组合、复方产品不适用于BE豁免 要求。
第V.D章节,指南草案和指南都声明了:本指南不适用于舌下片、含片。指南草案列出了5种药品(digoxin, lithium, phenytoin, theophylline, warfarin),指南定稿删除了列举,避免争议。
【导读专家:Garth Boehm博士】
识林® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com
岗位必读建议:
注册专员 :需熟悉生物等效性研究的要求,以确保注册申请文件的完整性和合规性。临床研究协调员 :应理解生物等效性研究设计、执行和评估的标准,以协调临床研究顺利进行。质量保证专员 :必须掌握生物等效性研究的质量控制和保证要求,确保研究符合GMP标准。研发人员 :需了解生物等效性研究的科学基础和方法,以指导药品开发过程。文件适用范围:
文件要点总结:
生物等效性研究设计 :强调了生物等效性研究设计的重要性,包括研究设计、参考和测试产品的选择、受试者、研究执行、特性调查、剂量强度、生物分析方法和评估方法。
生物等效性研究的法律基础 :明确了生物等效性研究的法律依据,包括适用的指令和法规。
生物等效性研究的执行 :规定了生物等效性研究执行过程中的标准操作程序,包括受试者的选择、研究条件的标准化、采样时间、禁食或餐后条件等。
生物等效性研究的评估 :强调了对生物等效性研究结果的评估方法,包括药代动力学参数的分析和接受限值。
生物等效性研究的报告 :要求提供完整的生物等效性研究报告,包括研究的详细文档、协议、执行和评估。
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
研发(R&D) :应深入理解生物药剂学分类系统(BCS)及其对药物开发的影响,确保新药设计符合FDA的生物利用度和生物等效性研究豁免标准。注册(Regulatory Affairs) :熟悉BCS分类及生物豁免(Biowaiver)的申请流程和要求,为IND、NDA和ANDA等申请提供法规支持。质量管理(QA) :掌握BCS分类标准,确保产品质量控制符合FDA指南要求,特别是在药物溶出度和稳定性方面。临床(Clinical) :了解BCS分类对临床试验设计的影响,特别是在生物等效性研究中。文件适用范围:
文件要点总结:
BCS分类标准 :明确了药物根据水溶性和肠道渗透性分为四类,为药物开发和生物豁免提供科学框架。生物豁免条件 :对于BCS 1类和3类药物,若满足快速或非常快速溶出,且辅料不影响药物吸收,则可能豁免体内生物利用度和生物等效性研究。药物溶出度测试 :强调了溶出度测试的重要性,包括测试条件、方法和相似度评估。辅料影响考量 :讨论了辅料对药物吸收可能产生的影响,并对BCS 1类和3类药物的辅料使用提出了具体要求。特定情形下的豁免限制 :对于窄治疗指数药物和设计用于口腔吸收的产品,不适用BCS基础的生物豁免。以上仅为部分要点,请阅读原文,深入理解监管要求。
法规指南解读 适用岗位(必读) 研发(R&D) :负责药品配方和工艺的开发,需了解成分和组成变更的级别分类。生产(Production) :涉及药品生产规模变更,需遵循相应的测试和文件要求。质量保证(QA) :确保所有变更符合监管要求,负责审核相关测试和文件。注册(Regulatory Affairs) :负责提交变更文件和维护药品注册状态。工作建议 研发 :在进行成分或组成变更时,应根据变更级别选择合适的测试和文件。生产 :在规模变更时,确保遵循CGMP并进行适当的验证。QA :审核所有变更文件,确保符合监管要求,并监督测试的执行。注册 :了解变更的监管路径,准备并提交相应的补充申请或年度报告。适用范围 本文适用于美国FDA监管下的化学药品的立即释放固体口服剂型。涉及新药申请(NDA)、简化新药申请(ANDA)和简化抗生素申请(AADA)的变更,包括成分或组成、生产地点、生产规模变更以及生产过程和设备变更。
文件要点 变更级别定义 :根据对药品质量和性能影响的可能性,将变更分为三个级别,并为每个级别提供测试和文件要求。化学、制造和控制测试 :明确了每个变更级别的推荐测试,包括稳定性测试和溶出度测试。生物等效性研究 :对于可能显著影响药品质量和性能的变更(Level 3),要求进行全面的生物等效性研究。文件提交要求 :根据变更级别,规定了年度报告、生效中变更补充申请和预先批准补充申请的提交要求。监管路径 :提供了基于21 CFR 314.70(a)的变更通知的简化途径,允许在提交补充申请时或在下一个年度报告中通知变更。以上仅为部分要点,请阅读原文,深入理解监管要求。