首页
>
资讯
>
【警告信和483】近期重点缺陷概要,以及用识林 AI 看483
出自识林
【警告信和483】近期重点缺陷概要,以及用识林 AI 看483
2025-07-26
药监部门检查报告是掌握GXP合规要求和趋势的重要学习资料,不仅包含合规“红线”供药企“有则改之”,还提示应避免的错误解决思路,应采取的正确方式。
识林目前收录4种检查报告,包括FDA警告信(药品警告信均提供人工翻译)和483,欧盟缺陷信,以及WHO的公共检查报告。
此外,识林还将警告信和483分别整理成数据库,基于国别、产品类型、检查官、缺陷关键词,法规条款等多个维度。
*识林免费用户可查阅警告信等检查报告原文,会员可查阅双语版本和“警告信数据库” 、“483数据库” 。
*会员可点击“帮助中心” 获得更多识林使用技巧。
近期更新的重点483和警告信及其缺陷概要
*文末还有如何使用识林AI看警告信和483的心得,分享给识林会员。
【警告信】Daewoo Pharmaceutical Co., Ltd.
FDA在对Daewoo Pharmaceutical Co., Ltd.的检查中发现多项CGMP违规行为。关键问题包括未建立和遵循防止微生物污染 的书面程序、无菌 和灭菌 过程验证不足;烟雾研究显示无菌处理操作中缺乏单向气流;培养基灌装程序未能准确模拟商业操作;无菌处理区域存在不良实践;人员监控不足以保证无菌性;生产和过程控制操作未在足够大小的特定区域内进行;质量控制单位未能确保产品符合CGMP和既定质量标准。FDA要求公司进行风险评估、整改计划、确保适当的无菌实践和洁净室行为、回顾性审查和风险评估、烟雾研究和培养基灌装程序的全面总结。此外,公司未能建立足够的清洁和维护设备书面程序。
【483】NATCO Pharma Limited
FDA在对NATCO Pharma Limited的检查中发现多个关键问题。首先,无菌处理区域的环境监测系统存在缺陷,特别是在非活性颗粒监测和数据完整性方面。例如,电子数据与官方报告的纸质结果之间存在差异,且在2019年7月至2024年7月期间,操作员在没有适当记录的情况下保存数据,可能导致数据操纵。其次,设备和器具的维护不当,可能导致产品污染 。此外,无菌工艺 的验证不充分,特别是在气流模式的可视化测试中。还有,未对无菌产品进行充分的微生物污染控制程序的建立和遵循,包括未对所有员工进行100%目检 的资格认证。最后,质量控制部门未能充分记录和审查色谱分析中的异常峰值,以及环境监测活动的记录不完整。
【483】Sun Pharmaceutical Industries Ltd.
FDA在对Sun Pharmaceutical Industries Ltd的检查中发现多项违规行为,主要涉及无菌操作和产品质量控制。首先,公司未能建立和遵循防止微生物污染的程序,如无菌产品的灌装 过程中存在不当操作和环境监控不足。其次,无菌工艺验证不足,培养基灌装试验未能准确模拟实际生产过程。此外,公司未能彻底调查生产过程中的异常,特别是在环境和人员监控出现偏差时。公司还未能确保药品生产设备 的设计和维护能够防止污染和故障,例如稳定性试验室天花板裂缝导致液体滴落,可能影响产品质量。最后,批生产和控制记录不完整,无法确保生产和控制的准确性和完整性。
【483】Meds For Vets
Meds For Vets公司在FDA检查中被发现存在多项违规行为,涉及无菌工艺验证、设备管理、质量控制 、人员操作和稳定性 测试等方面。具体问题包括未校准的气压表、未经验证的灭菌设备 、未使用生物指示剂 、无菌药品目检无标准操作规程、使用非药用辅料、容器灭菌与除热原处理不当、设备适用性不足、生物指示剂使用不当、人员操作污染、洁净区 管理不当、培养基灌装试验缺陷、质量控制失效、烟雾研究不充分以及测试与稳定性程序缺失。
【483】STAQ Pharma of Ohio, LLC
STAQ Pharma of Ohio, LLC在FDA检查中被发现存在多项违规行为。首先,公司未能彻底审查批次不符合规格的情况,特别是在盐酸右美托咪定注射液的100%目检和AQL检查中发现的关键缺陷,公司未进行根本原因调查即放行该批次,且未对发现的颗粒物进行特性分析或采取纠正措施。其次,公司未能建立充分的书面生产和过程控制程序,抽样检查和目检程序不明确,缺乏对不同缺陷类别的接受标准和趋势分析。最后,公司药品标签未包含FDCA要求的活性成分信息,违反了法律要求,可能影响患者用药安全。
【483】New England Home Therapies, Inc.
New England Home Therapies, Inc.被查出的主要缺陷包括人员着装不符合ISO 5区域的无菌要求,包括无菌服、面罩和发罩。化学防护服未达到无菌标准,且人员在穿戴无菌手套前未进行手部消毒 。无菌药品的微生物污染预防程序未建立或未遵循,包括药品生产用品未在进入ISO 5区域前进行消毒,以及人员在无菌操作时手臂直接接触设备和组件。设备和工具的维护未能防止药品污染,影响药品的安全性、身份、效力、质量和纯度,如HEPA过滤器上的污渍未被记录或评估。无菌处理区域的环境监测系统存在缺陷,包括空气和表面监测结果不能代表常规操作条件,以及压差监测不频繁,等等。
【483】Fusion IV Pharmaceuticals, Inc. dba Axia Pharmaceutical
Fusion IV Pharmaceuticals, Inc. dba Axia Pharmaceutical在无菌药品生产过程中存在多项不符合规定的情况。首先,公司未能建立防止微生物污染的程序,特别是在ISO 5级别的层流工作站中进行的培养基灌装无菌性失败未进行根本原因调查。其次,公司未对无菌药品的灭菌过程进行充分验证,包括灭菌设备的安装和操作确认以及性能确认。此外,公司未能有效控制内毒素 和微生物负荷,且在性能确认活动中记录不全。公司还未能对无菌处理区域的环境条件进行适当监测,缺少对“非批次”产品的每日环境监测。公司未对每批需要无菌的药品进行适当的实验室测试,包括方法适用性测试和抗菌效力研究。
【483】Apothecary Pharma LLC
FDA在对Apothecary Pharma LLC的检查中发现多项违规行为。首先,质量控制单元(QCU )未遵循书面程序,未能确保批记录的完整性,未审查关键的认证报告,且部分操作程序草案未最终确定。其次,生产过程中员工未穿戴适当的防护服,违反了清洁室出入程序,增加了产品污染风险。此外,无菌处理区域在设备维护、清洁消毒方面存在缺陷,影响无菌条件的维持。FDA还指出,公司未进行足够的烟雾测试以验证无菌和灭菌过程的有效性,且生产控制程序未能有效防止微生物污染。最后,公司未向FDA提交所需的产品报告,违反了503B条款 的规定。
如何与识林AI聊聊警告信和483?
专业而细心的识林读者可能已经发现,上述内容主要来自识林AI的解读。
警告信和483包含大量文字内容,而483往往是扫描件pdf的格式,且有部分信息被涂抹,更不易读。
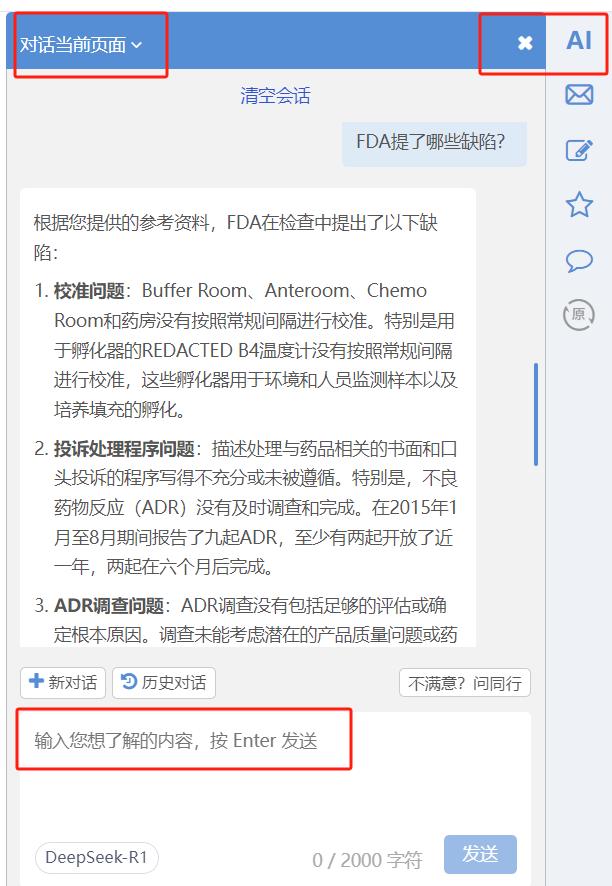
识林已经将AI接入了每个警告信或483页面。识林会员可点击页面右上角的“AI”按钮,激活AI对话页面,选择“对话当前页面”(如下图红框),此时这份文件已被传递给AI,这个窗口就成了一位熟读这份警告信或483、且专属于你的SME(主题专家),与你开展对话。
你可以先笼统地问“FDA提了哪些缺陷?”把握全文重点,例如:
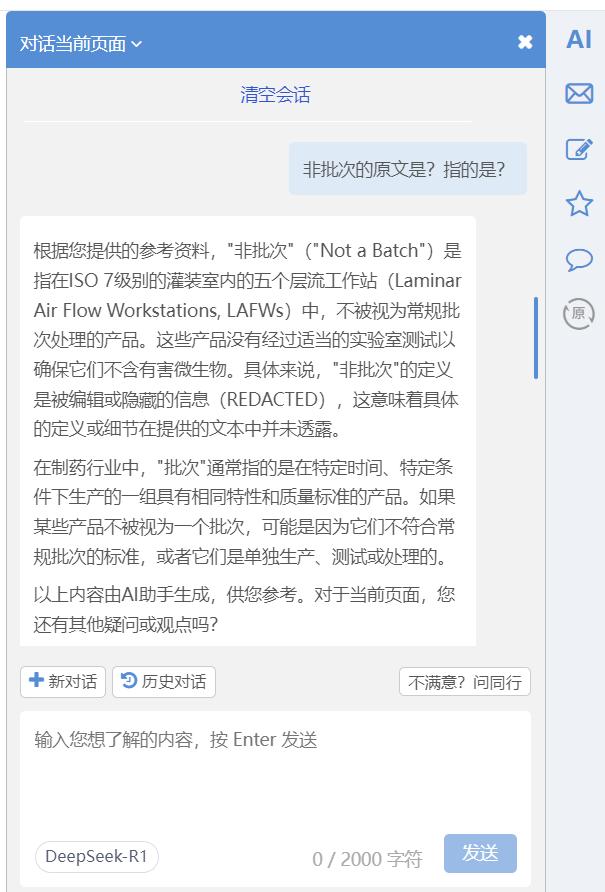
也可以基于AI的回答进一步追问,例如对上述Fusion IV 的483中所谓的“非批次”追问“非批次的原文是?指的是?”:
AI虽然不可尽信,仅供参考和支持,但识林AI配合知识库,不仅更便捷,也可能更“靠谱”,欢迎识林会员一起参与探索。
识林® 版权所有,未经许可不得转载
适用岗位及工作建议:
QA(质量保证):必读。需审查和更新质量控制单元(QCU)的书面程序,确保批记录的完整性和合规性。 生产(Production):必读。应强化无菌操作培训,确保员工正确穿戴防护服,遵循清洁室出入程序。 注册(Regulatory Affairs):必读。负责向FDA提交所需的报告,包括初始产品报告和503B外包设施注册。 文件适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
QA(质量保证):必读。需根据FDA 483观察结果,审查和更新无菌药品生产和质量控制流程,确保微生物污染控制措施得到有效实施。 生产(Production):必读。应重新评估无菌生产操作,特别是在ISO 5环境下的行为规范,以及容器和密闭系统的灭菌处理。 研发(R&D):必读。需要检查产品开发过程中的微生物污染控制措施,确保产品在研发阶段就符合FDA要求。 注册(Regulatory Affairs):必读。必须根据FDA 483中的观察项更新注册文件和申报资料,确保所有产品标签和报告符合FDA规定。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
QA(质量保证):必须阅读,以确保所有无菌和非无菌药品的生产过程符合FDA的监管要求,特别是无菌工艺验证和微生物污染防控。 生产(Production):必须阅读,以确保生产过程中使用的设备和方法得到适当验证,并且操作符合无菌操作标准。 QC(质量控制):必须阅读,以确保药品的测试和放行符合规定,包括活性成分的鉴定和效力测试。 注册(Regulatory Affairs):必须阅读,以了解FDA的观察和要求,以便制定和实施纠正措施。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
QA(质量保证部门):必读。建议全面审查和更新SOPs,特别是与无菌操作和数据完整性相关的程序,确保所有电子数据得到适当审查和存档。 生产(Production):必读。建议对无菌生产区域进行维护和升级,以符合GMP要求,确保设备和环境监控符合规定标准。 研发(R&D):必读。建议对无菌工艺进行再验证,确保工艺的微生物控制措施得到充分验证。 市场(Marketing):必读。建议评估市场影响,准备应对可能的产品召回或市场限制。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):必须确保所有操作符合FDA规定,特别是无菌药品的生产过程。 生产(Production):需要根据观察结果调整无菌药品生产流程。 环境监测(Environmental Monitoring):负责改进环境监测程序,确保无菌条件。 工作建议:
QA:审查和更新SOPs,确保所有操作符合FDA的最新要求,特别是无菌操作和环境监测。 生产:立即整改观察中提到的问题,如人员着装、无菌操作程序等,并进行再培训。 环境监测:重新评估和设计环境监测计划,确保能够代表常规操作条件,并覆盖所有关键区域。 适用范围:
文件要点总结:
人员着装不符合ISO 5区域的无菌要求,包括无菌服、面罩和发罩。化学防护服未达到无菌标准,且人员在穿戴无菌手套前未进行手部消毒。 无菌药品的微生物污染预防程序未建立或未遵循,包括药品生产用品未在进入ISO 5区域前进行消毒,以及人员在无菌操作时手臂直接接触设备和组件。 设备和工具的维护未能防止药品污染,影响药品的安全性、身份、效力、质量和纯度,如HEPA过滤器上的污渍未被记录或评估。 无菌处理区域的环境监测系统存在缺陷,包括空气和表面监测结果不能代表常规操作条件,以及压差监测不频繁。 未能彻底审查任何未解释的差异,无论批次是否已分发,缺乏对失败和差异的充分书面调查。 无菌药品的微生物污染预防程序未包括灭菌过程的验证。 机械和电子设备的常规校准未根据书面程序执行,关键设备未定期校准。 药品投诉处理程序书写不当或未遵循,ADR调查不及时,未充分评估或确定根本原因。 以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
QA(质量保证):必读。需审查和更新无菌操作和环境监控程序,确保符合FDA规定。 生产(Production):必读。应加强员工无菌操作培训,改进生产流程,减少微生物污染风险。 研发(R&D):必读。在新药开发过程中,需关注无菌工艺验证和产品质量标准。 文件适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):必读。需评估和更新质量控制流程,确保符合CGMP规定。 生产(Production):必读。需根据CGMP要求改进无菌操作和设备管理。 研发(R&D):必读。需确保产品开发符合CGMP规定,特别是在无菌处理方面。 质量控制(QC):必读。需改进原料和成品的检测流程,确保产品质量。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
外包设施运营人员(Outsourcing Facility Operators):必须了解注册和报告要求,确保设施符合规定。 法规事务专员(Regulatory Affairs Specialists):应熟悉相关定义和法规,指导合规运营。 质量保证专员(QA):需监督药品标签和包装要求,确保符合规定。 文件适用范围:
文件要点总结:
注册与报告要求 :外包设施需每年注册并报告药品信息,包括活性成分来源等详细数据。药品成分规定 :禁止使用未列入临床需求清单或药品短缺清单的原料药,除非满足特定条件。标签与包装标准 :药品标签必须包含特定信息,如“这是一个复合药品”的声明和外包设施的联系信息。风险评估与缓解策略 :对于需要风险评估和缓解策略的药品,外包设施必须展示相应的控制措施。禁止批发销售 :禁止除复合药品的外包设施外的其他实体销售或转让药品。以上仅为部分要点,请阅读原文,深入理解监管要求。