|
首页
>
资讯
>
能力评估 无菌保证随堂测试
出自识林
2024-03-08
以下文章来源于IPEM ,作者IPEM
无菌保证是无菌制造过程的核心关注点,贯穿于产品、工艺、方法、厂房、设施、设备、物料、人员、运输、贮存和使用等所有相关系统。作为IPEM核心系列课,将以解决无菌制造实际问题为目的,系统全面讲解与无菌产品相关的历史、理念、法规、工艺、设施设备、监测、验证和风险管理,阐明无菌保证的相关理念、逻辑、体系和实践。本月16-17日,尹放东博士与多位礼来(苏州)老师将继续和大家分享无菌保证的内容“无菌保证(一)”。
同时,为评估学员对知识点的掌握、夯实学习效果,今后IPEM将针对每次课程进行随堂测试,分析测试结果后,为每位学员准备个人评测报告,为企业会员准备团队评测分析报告。本文将以去年8月“无菌保证、生物工艺开发和验证”学员们随堂测试结果为样本案例进行分析,欢迎大家就分析角度和方法提出建议,同时也为即将报名听课的企业和同学提供参考。
基于制药岗位知识结构的考核体系
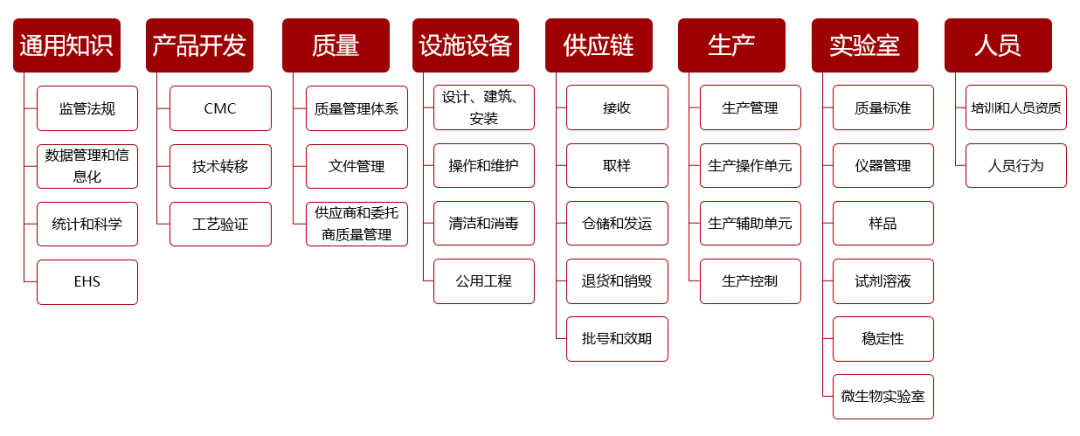
结合IPEM多年教学经验和国际机构发布的产业岗位能力和知识管理案例,经过与美国FDA和跨国药企资深培训管理人员的深入讨论,IPEM与识林团队共同开发了一套基于岗位的知识结构体系。
该结构从产品生命周期出发,将制药知识细分为八大类别:通用知识(包括注册合规)、产品开发、质量、设施设备、供应链、生产、实验室和人员。在这八大一级知识点下,又细分为二级和三级知识点。这些知识点对不同岗位的重要程度不同,基于此,最终为不同岗位建立起各自的岗位知识结构,包括:
- 核心知识:各岗位日常工作需要掌握的知识点,和岗位最为相关;
- 重要指示:需要了解的知识点,有助于频繁的跨部门沟通;
IPEM和识林团队以官方机构发布的法规、指南、指导原则等文件为基础,整理了超过4000道试题,并精确匹配到相应的知识点。通过系统化的考试组织,对学员能力进行量化评估。目前,已有恒瑞、科伦、齐鲁、华润、扬子江等多家企业参与,覆盖了QA、QC、生产、工艺开发、工程设备等多个岗位,积累了超过2000人次的测试数据。
考了哪些知识点?学员们考得怎么样?
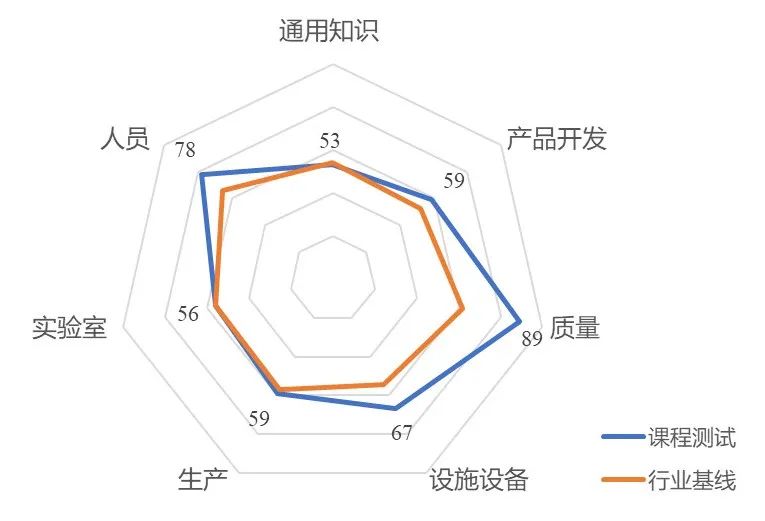
在测试中,每位学员随机抽取20道题进行作答。本次共有64位学员参与了随堂测试,平均成绩为65分。题目覆盖:无菌工艺模拟试验(APS)、屏障技术(隔离器和RABS)、污染控制策略(CCS)、灭菌工艺及验证、除菌过滤等。从知识点分布来看,本次测试覆盖了通用知识(注册合规)、产品开发、质量、设施设备、生产、实验室和人员七大知识点。与行业基线(恒瑞、科伦、齐鲁等多个岗位的测试数据)相比,本次测试在产品开发、质量、设施设备、人员方面的得分均高于基线,而在通用知识(注册合规)、生产和实验室方面的得分与基线持平。
具体来看,各三级知识点(测试题量≥40人次)的得分情况如下:
表1 各项知识点得分
| 知识点
| 平均分
|
| 质量 → 质量管理体系 → 质量管理的目标、元素、职责和实施
| 86
|
| 人员 → 人员行为 → 无菌生产区操作准则和人员监测
| 78
|
| 设施设备 → 操作和维护 → 设施设备使用
| 75
|
| 设施设备 → 设计、建筑、安装 → 洁净级别的设计、确认和维护
| 70
|
| 生产 → 生产控制 → 污染和交叉污染
| 68
|
| 产品开发 → 工艺验证 → 无菌工艺验证
| 61
|
| 生产 → 生产操作单元 → 关键单元操作
| 59
|
| 设施设备 → 清洁和消毒 → 灭菌
| 57
|
| 生产 → 生产控制 → 环境控制
| 57
|
| 生产 → 生产管理 → 原辅料和组分流转控制
| 52
|
| 生产 → 生产控制 → 中间过程控制
| 52
|
从随堂测试结果来看,灭菌工艺及其验证、生产环境控制、物料转运、中间过程控制等知识点得分相对偏低,后续无菌保证课程将对此强化,同时建议参加后续课程的学员提前预习相关内容。
易错题分析
1. 对于最终灭菌产品,企业可根据自身情况制定灭菌前药液的微生物负荷标准,下列描述错误的是:(单选题)
- a. 无论采用过度杀灭法还是残存概率法的灭菌工艺,均应对每一批药液灭菌前的微生物污染水平进行监测
- b. 原则是确保每个容器在灭菌后无菌保证水平达到 10^-6的水平
- c. 微生物负荷标准的制定应有依据和理由,常见的如每 100ml 药液中污染菌不超过100cfu
- d. 通常灌装后段配制药液储存的时间最长,微生物滋生的风险增加,应对灌装后段产品进行取样,监测微生物污染水平
解析:答案为a。
该题的正确率为29%,多数错误选择了c选项。这道题考核的是对灭菌原理的理解。
在恒定的热力灭菌条件下,同一种微生物的灭活符合一级动力学方程(也叫存活曲线),微生物死亡速率是微生物耐热参数D和杀灭时间的函数。即在给定的时间下被灭活的微生物与仍然存活数成正比:
- LgNt=IgN0 - F(T,Z)/DT
- Nt:t分钟后微生物计数值
- N0:初始微生物计数值
- DT:在T温度下,微生物降低一个对数单位所需要的时间(单位:分钟)
- F(T,Z):灭菌程序在确定温度系数Z的T温度的等效灭菌时间
课程中对湿热灭菌程序进行了介绍,包括过度杀灭法和残存概率法:
- 过度杀灭法的目标是确保达到一定程度的无菌保证水平,而“不管被灭菌品微生物初始数量及其耐热性如何”,该方法对初始菌数量及耐热性作了最坏的假设,因此从产品无菌保证水平角度看,对被灭菌品不需要进行常规的初始菌监控;
- 残存概率法的设计要考虑初始微生物的分析,另外加上安全系数,耐热性的设定值仅比实际检出微生物的耐热性稍高,微生物污染水平的增加或耐热性增加都会造成灭菌程序的目标失败,因此应考虑监控产品或单个被灭菌容器的初始微生物污染水平、检出菌耐热性;
这道题是选择描述错误的选项。对于a选项,其描述错误的点在于:过度杀灭法对初始菌数量及耐热性作了最坏的假设,不需要进行每批的微生物污染水平监测。对于c选项,其描述是正确的,100cfu/100ml是常用的最终灭菌产品灭菌前微生物负荷监测标准。有很多学员选择c选项,可能是把非最终灭菌产品(如过滤除菌工艺)和最终灭菌产品的微生物负荷标准弄混了:在EMA发布的Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container中,建议最终灭菌产品灭菌前微生物负荷标准为100cfu/100ml,过滤除菌产品则为10cfu/100ml。
| 本题考核知识点:生产 → 生产控制 → 中间过程控制
该知识点与不同岗位的关联程度如下:
工艺开发:为岗位重要知识点,需了解;
QA:为岗位重要知识点,需了解;
QC:为岗位相关知识点,仅需知道;
生产:为岗位重要知识点,需了解;
|
2. 关于RABS或隔离器手套进行完整性测试错误的是:(单选题)
- a. 对于RABS手套,应在每Batch 或 Campaign前和后进行完整性检测,每次使用时或当有影响完整性时目检
- b. 测试方法目前没有强制的法规规定,不同设备厂家的手套测试方有着不同的方法,企业在使用该方法时需要经过合适的评估和验证
- c. 完整性测试方法举例:将手套快速加压至1200pa(使手套能够完全伸展开),上下震荡稳定后,保持10分钟,压差不得低于1100pa为合格
- d. 手套完整性测试的方法需要经过验证,例如使用标准的手套测试针(举例:100μm)对手套进行穿刺,测试是否能够被检出
解析:答案为a。
该题的正确率为35%,多数错误选择了d选项。这道题考核的是屏障技术相关的使用要求。
屏障技术是中国和PIC/S(欧盟/WHO)无菌附录有差异的主要内容之一,同时也是无菌保证课程中介绍的重点。这道题参考的是PIC/S无菌附录的4.21条:
4.21 应证明手套系统(用于隔离器和RABS)的材料具有适当的机械和化学抗性。更换手套的频率应在CCS中定义。
i. 隔离器:
a. 对于隔离器,手套系统的检漏测试所用的方法应经过证明适用其任务和关键程度。应定期进行测试。通常,手套完整性测试至少应在每个批次或阶段性生产的开始和结束时进行。根据经过验证的阶段性生产长短,可能需要进行额外的手套完整性测试。
……
ii. RABS:
对于RABS,A级区使用的手套应在安装前进行灭菌,并在每次生产活动之前通过经验证的方法进行灭菌或有效的生物去污染。如果在操作过程中暴露于背景环境中,应在每次暴露后使用经批准的方法完成消毒。应在每次使用时目视检查手套,并定期进行完整性测试。
这道题是选择描述错误的选项,对于选项a,“应在每Batch 或 Campaign前和后进行完整性检测”的要求是针对隔离器的,对于RABS,这种检测要求是过高的:虽然隔离器和RABS都是通过将A级环境与周围房间环境分隔来提供保护,但隔离器在密封性上的要求远高于RABS,这就导致隔离器的泄漏风险要远高于RABS,基于此,隔离器对手套的完整性测试的要求就更为严苛。
对于c和d选项,是手套完整性测试和测试方法验证的两个举例,这部分并没有法规要求,企业使用相关方法时应经过适当的评估。
| 本题考核知识点:设施设备 → 操作和维护 → 设施设备使用
该知识点与不同岗位的关联程度:
工艺开发:与该岗位不相关;
QA:为岗位相关知识点,仅需知道;
QC:与该岗位不相关;
生产:为岗位核心知识点,需掌握;
|
新一期课程,内容升级和强化体系
无菌保证系列2024年将设计为四次课程:
第一课主要介绍无菌产品的历史起源,探讨无菌保证的基础、理念和法规;污染控制策略 (CCS) 的核心理念和法规要求,污染控制策略文件的合理设计和合规撰写,并且深入的探讨污染控制策略在无菌生产领域的实施和应用:无菌工艺模拟试验。
后续三次课程将进一步探讨和分享其他无菌保证领域的主题和热点、法规难点和行业痛点。报名请点链接或扫描下方二维码。
法规指南解读 适用岗位: 工作建议: - QA:确保所有生产活动符合GMP要求,监督无菌药品生产流程。
- 生产:按照GMP要求执行无菌生产操作,确保产品质量。
- 研发:在药品开发阶段考虑GMP合规性,设计符合要求的生产流程。
- 临床:确保临床试验用药的无菌性和质量符合GMP标准。
- 注册:在药品注册过程中提供符合GMP要求的生产和质量控制信息。
适用范围:
本文适用于化学药品、生物制品的无菌药品生产,包括原料药、制剂等。适用于欧盟地区的Biotech、大型药企、跨国药企等。 要点总结: - 无菌药品生产环境:强调了对无菌药品生产环境的严格控制,包括洁净室的分类和设计,以及对生产环境的持续监测。
- 质量风险管理(QRM):在整个文件中,QRM是确保无菌药品生产质量的核心原则,要求企业在设计和控制生产设施、设备、系统和程序时应用。
- 关键控制点:提出了无菌药品生产过程中的关键控制点,包括设施设计、设备操作、过程验证、环境监测和人员培训等。
- 污染控制策略(CCS):强调了CCS在无菌药品生产中的重要性,要求企业实施全面的CCS以确保产品质量和安全。
- 无菌工艺验证:要求对无菌工艺进行验证,包括无菌过程模拟(APS)和其他相关测试,以确保生产过程能够持续产生无菌产品。
以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读适用岗位(必读)- QA:确保无菌药品生产全过程符合GMP要求。
- 生产管理:负责无菌药品生产的具体操作和管理。
- 设备维护:保障生产设备符合洁净度要求。
- 研发:涉及无菌药品的研发流程和工艺设计。
工作建议- QA:定期审查生产流程,确保符合附录1规定。
- 生产管理:制定和执行无菌操作规程,监控生产环境。
- 设备维护:定期验证和维护空气净化系统及其他关键设备。
- 研发:在产品设计阶段考虑无菌生产要求,优化工艺。
适用范围本文适用于无菌制剂和无菌原料药的生产,包括化学药和生物制品,特别针对无菌药品的GMP生产要求。适用于在中国进行无菌药品生产的企业,包括Biotech、大型药企、跨国药企等。 文件要点总结- 无菌药品定义及分类:明确无菌药品包括无菌制剂和无菌原料药,分为最终灭菌产品和非最终灭菌产品。
- 生产原则:强调无菌药品生产应最大限度降低污染,且不应仅依赖最终处理或成品检验。
- 洁净度级别及监测:规定了洁净区的级别划分、悬浮粒子和微生物的监测标准和方法。
- 隔离操作技术与吹灌封技术:对高风险操作的隔离操作和吹灌封技术提出具体要求。
- 灭菌工艺与方法:详细规定了湿热灭菌、干热灭菌、辐射灭菌、环氧乙烷灭菌和过滤除菌的方法和要求。
结语以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读 适用岗位: 工作建议: - QA:确保所有生产活动符合GMP要求,监控生产过程,确保无菌操作符合规定。
- 生产:遵守无菌操作规程,执行环境和设备监测计划。
- 设备维护:确保设备维护不引入污染,维护后进行必要的清洁和灭菌。
- 环境监测:定期进行环境监测,确保洁净室符合规定的洁净等级。
- 微生物实验室:进行必要的微生物测试,包括无菌检验和微生物限度检查。
文件适用范围:
本文适用于无菌药品的生产,包括化学药品、生物制品和疫苗等。适用于原料药、辅料、内包装材料和成品制剂的无菌生产。适用于采用无菌工艺和最终灭菌工艺的产品。发布机构为PIC/S,适用于跨国药企和大型药企。 要点总结: - 无菌药品生产环境:强调了对无菌药品生产环境的控制,包括洁净室的分类和设计,以及对环境监测的具体要求。
- 风险管理:提出了在整个生产过程中应用质量风险管理(QRM)的原则,以识别、评估和控制潜在的质量风险。
- 污染控制策略:要求制定污染控制策略(CCS),以界定关键控制点并评估所有控制措施的有效性。
- 无菌工艺模拟:对无菌工艺模拟(APS)提出了具体要求,以验证无菌工艺的有效性。
- 质量控制:强调了质量控制的重要性,包括对原辅料、中间产品和成品的微生物、微粒和内毒素/热原的控制。
以上仅为部分要点,请阅读原文,深入理解监管要求。 WHO无菌制剂的药品生产质量管理规范(GMP)解读 适用岗位: - QA(质量保证):负责确保生产过程符合GMP要求。
- 生产:涉及无菌制剂生产的各个环节。
- 工程:维护和验证生产环境和设备。
- 微生物实验室:进行环境监测和产品测试。
工作建议: - QA:定期审查生产流程,确保符合GMP规范。
- 生产:严格遵守无菌操作规程,进行生产活动。
- 工程:确保生产环境和设备得到适当维护和验证。
- 微生物实验室:准确进行环境和产品微生物测试,及时报告数据。
文件适用范围:
本文适用于化学药、生物制品、疫苗和中药等无菌制剂的生产企业,包括创新药、仿制药、生物类似药和原料药等注册分类。适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业类别,由WHO发布。 要点总结: - 质量风险管理:强调在所有生产环节应用质量风险管理原则,以预防微生物、颗粒物和内毒素/热原污染。
- 生产环境和设备:规定了对生产环境和设备的设计与验证要求,包括洁净室、隔离技术和设备。
- 人员资质与培训:要求生产人员具备适当的资格、经验和培训,以确保在生产、包装和分销过程中保护无菌产品。
- 生产过程控制:涉及无菌制剂的特定技术,如终端灭菌产品、无菌制备和加工、无菌产品的完成处理等。
- 环境和过程监控:包括环境监测的一般要求、总颗粒物监测、活性颗粒物监测和无菌工艺模拟。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。
- 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。
- 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。
适用范围:
本文适用于涉及化学药、生物制品、疫苗和中药等药品类型的企业,包括创新药、仿制药、生物类似药和原料药等注册分类。适用于不同规模的企业,如Biotech、大型药企、跨国药企、CRO和CDMO等,由相关药品监管机构发布。 文件要点总结: - 质量管理体系概述:明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。
- 产品质量实现要素:涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。
- 质量保证要素:包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。
- 质量风险管理:介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。
- 质量管理系统文件:规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |