PDA在2016年推出“数据可靠性行为守则要素(Elements of a Code of Conduct for Data Integrity)”的指南,2018年7月又出版了“PDA 讨论要点:文档/数据管理与控制以及数据可靠性迎检准备的最佳实践(PDA Points to Consider: Best Practices for Document/Data Management and Control and Preparing for Data Integrity Inspections)”【不要光从错误中学习 — PDA发布数据可靠性最佳迎检实践问答】,8月中旬,PDA发布了技术报告80,制药实验室的数据可靠性管理体系(Data Integrity Management System for Pharmaceutical Laboratories),这份长达56页的报告中,覆盖了原始记录(4.1节),人因(4.2节),混合记录体系(6.2节),计算机系统(6.3节),数据管理软件(6.5节)、电子数据管理(6.6节)、数据管理体系的风险管理(7章)和数据可靠性问题的整改(8章),提供了很多实施细节和案例,第五章微生物实验室是特色,尽管其它法规指南也有涉及相关内容,但系统梳理微生物实验室的数据可靠性风险是该报告的亮点,下面识林以“5.2环境监测、无菌和内毒素检验”为例,解析该报告。
5.2.1 Environmental Monitoring Equipment 环境监测装置
Some of the equipment used most frequently in the pharmaceutical microbiology laboratory and manufacturing facility are environmental monitoring (EM) devices used to collect volumes of air or surface contact from specific rooms (e.g., ISO-5 manufacturing areas used for filling sterile products or sterility test suite used to analyze finished drug products labeled as sterile). The neglect or mishandling of EM devices can cause misleading or incorrect analytical results about the microbial presence (bioburden) within the room being monitored.
在微生物实验室和生产设施中最常用的一些设备是用来收集特定房间的大量空气或表面接触物的环境监测(EM)装置(例如,灌装无菌产品的ISO-5生产区或用于分析标明无菌的成品的无菌检验单元)。对EM设备的疏忽或误操作可能导致被监测房间关于微生物存在(生物负荷)的误导或不正确的分析结果。
5.2.2 Air Sampling Devices 空气取样装置
In general, an air sampling device requires that a petri dish or a specifically designed plastic strip containing nutrient agar be placed into the device; the instrument is then turned on to draw a specific volume of air into contact with the agar surface. At the end of the air sampling dwell time, the petri dish or strip along with any captured microorganisms are removed from the device and placed in an incubator. Subsequently, the number of growing microorganisms on the media is counted. Examples of where data integrity problems can occur with air sampling devices include:
通常,空气取样装置需要将一个培养皿或特殊设计的含有营养琼脂的塑料条放在装置中;打开装置以吸取特定体积的空气与琼脂表面接触。空气取样停留时间结束时,将培养基或塑料条连同捕获的微生物一起从装置中转移至培养箱中。随后,将培养基上生长的微生物进行计数。空气取样装置可能出现数据可靠性问题的示例包括:
Visualization of the Petri Dish
培养皿的可视化
Signs of agar impingement and/or colonies growing within the agar medium, caused by the directed air being drawn into the device onto the agar surface, should be visible, ensuring that the agar petri dish was placed into the air handling device during the initial room setup or at another time, when appropriate.
由于空气直接被吸入到装置中的琼脂表面上,琼脂冲击和/或在琼脂培养基中生长的菌落的迹象应该是可见的,确保初始房间安装时或其他适当的时间琼脂培养皿被放在空气处置装置中。
识林解析:
促生长测试是关于培养基的数据可靠性风险的传统关注点,识林483数据库中至少收录了55个相关案例,但这里强调的,培养基被实际放置的证据是过去检查缺陷中不常出现的,可能引导今后的检查中,检查员开始关注培养基因空气采样而留下的痕迹。
此外,视频审核仍然这类问题暴露的关键(参见都是【都是视频惹的祸 — 从世界杯看视频回放的合规风险 2018-07-11】),在2016年03发给印度Emcure的警告信中提到: FDA检查员发现了许多不完备,不准确,或被假造的实验室记录的例子。 a. 无菌灌装区域里主动空气监测EM记录报告了已收集的样品,而实际上这些样品没有被采集过,有些文件记录报告了零CFU的EM结果,而实际上这些所谓的报告结果的样品根本都没有被采集过。FDA在检查期间审核过的同步录制视频记录显示这些EM样品并没有被采集过,即使该公司的实验室记录报告了这些样品的检测结果。FDA检查员发现该公司假造多个药品中未采集过的样品的EM结果,包括XX注射剂USP批号和XX注射剂批号XX。 尽管该公司的化验室里对这些药品和批次的记录显示收集了主动空气样品,FDA检查员在检查期间审核的视频证明操作人员实际上并没有取样。在检查期间,微生物化验员确认这些EM样品从来都没有被采集过。另外,有两个微生物化验员告诉检查员,这些培养皿有标签,放进去培养,弄的好像是真的在环境中暴露采过样似的。但是,这些培养皿确实从未暴露于环境中。但微生物化验员说因为“工作压力”一直都是这么做的。由于EM结果是假造的——仿佛取过样和/或并没有CFU生长,不能确保公司在此区域生产的注射剂药品在无菌灌装工艺结束时是无菌的。
Clogged Samplers
取样器堵塞
Over time, the narrow opening of the EM sampler that funnels the air directly toward the turning petri dish will become clogged, essentially preventing the full volume of air to impinge on the agar surface. Though a maintenance or operational anomaly, this directly impacts the data integrity of the microbial evaluation of critical work areas. Routine cleaning of the device in that narrow opening is essential to ensure data integrity for this equipment.
随着时间的推移,将空气直接漏向转动的培养皿的EM取样器狭窄开口会被堵塞,本质上阻挡全部体积的空气冲击琼脂表面。虽然是维护或操作异常,但这直接影响关键工作区域的微生物评估的数据可靠性。装置狭窄开口的常规清洁对于该设备的数据可靠性至关重要。
识林解析:
取样器的安装和位置导致监测数据不可靠是过去常见的缺陷,如,2014年04月发给墨西哥 Instituto Bioclon的警告信中: b. 企业的调查确认了,派去运行(b)(4)浮游菌空气取样器的分析员,没有正确安装和运行空气取样器,但仍用此进行标记和培养。2014年1月16日的培养皿目检,没有显示(b)(4)活性浮游菌空气取样器暴露的证据。
本指南中强调了一条非常具体的关注点:清洁维护不当导致取样器开口堵塞,这比正确安装的要求要高的多,不仅要有充分的验证维护规程和记录,可能还需要数据来支持。
Quality Control Logbook
质量控制日志
As with other laboratory equipment, recording if or when any of the air sampling devices are defective or out of service in the quality control logbook is essential. The dates during which a piece of equipment is listed as “out of service” would not be found in the sample batch records.
与其他实验室设备一样,若任一空气取样装置故障或停止运行时在质量控制日志中记录是至关重要的。设备的一部分被列为“停止运行”的日期没能在样品批记录中被发现。
沉降皿
When settle plates are used to passively monitor the manufacturing or laboratory environment, plates located near surfaces directly exposed to UV light during monitoring may not be suitable. UV light may kill viable airborne microbial contamination, resulting in questionable results. Settle plates exposed too long become desiccated, and unwrapped settle plates exposed within an isolator during vaporized hydrogen peroxide (VHP) or other decontamination steps become unable to support growth. In either case, potentially viable airborne microbial contamination may not be recovered during exposure.
当沉降皿用于被动监测生产或实验室环境时,在监测期间表面直接暴露于UV的位置附近放置的板子可能不合适。UV可能会杀死可空降微生物污染,导致可疑结果。暴露太久的沉降皿会变干燥,并且在过氧化氢(VHP)或其他消毒步骤期间暴露在隔离器中的打开的沉降皿变得无法支持生长。在任一情况下,在暴露期间可能无法复苏潜在可空降微生物污染。
最终计数
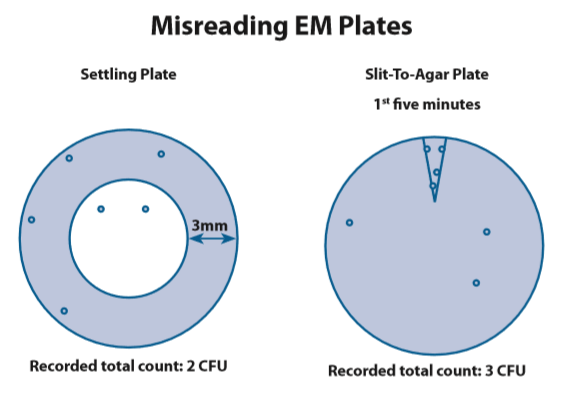
Any manipulation or adjustment of final microbial counts without sound scientific justification, such as subtracting colony counts near the edge of the petri plates or the number of bacterial colonies appearing at the beginning time period for air sampling plates, is a concern for data integrity (Figure 5.2.2-1).
在没有合理科学理由的情况下对最终微生物计数进行任何操作或调整,例如减去培养皿边缘附近的菌落计数或空气取样初期出现的细菌菌落数量,是数据可靠性问题(下图)
5.2.3 Surface and Personnel Monitoring Equipment 表面和人员监测设备
Disinfectants should not be applied to a work surface or on an operator’s gloved hands prior to sampling those surfaces using replicate organism detection and counting (RODAC) plates. Application of a disinfectant to a surface designated to be monitored before the test is performed will change the results of the assay, making the surface appear to be free of microorganisms or producing a lower bioburden. The manipulation of procedural requirements can obscure the reality of a contaminated facility or a person’s poor aseptic technique resulting in the manufacture of a drug product under conditions that adulterate the finished product and impact patient health. Data integrity detection may need to occur at microbiological recovery stages long before an analyst opens the incubator door and transfers the tubes and petri dishes for colonies to be counted, otherwise, the data may be compromised and meaningless. In some cases, the surface monitoring is not done correctly, e.g., too short a contact time or performed improperly (for instance, only the finger tips are sampled but the fingers are not correctly rolled over the agar, or fingers are overlapping the plate).</font>
在使用复制生物体检测和计数(RODAC)板对这些表面进行取样之前,工作表面或操作员戴着手套的手不应使用消毒剂。在进行检测之前对指定监测的表面使用消毒剂将改变检测结果,使表面呈现出没有微生物或产生较低的生物负荷。规程要求的操作可能会使设施污染或是人员不良无菌操作污染的事实变得模糊,导致在成品掺假和影响患者健康的条件下生产药品。在分析人员打开培养箱门将用于菌落生长的试管和培养皿进行计数之前,数据可靠性检验可能需要早在微生物复苏阶段开展,否则,数据可能会收到损坏和无意义。在某些情况下,表面监测做的不正确,例如,接触时间太短或执行不当(如,只有指尖被取样但手指没有正确的划过琼脂,或手指刨到了平板)。
5.2.4 Review of Sterility Test 无菌检验的审核
The laboratory methods employed to conduct a sterility test on finished sterile pharmaceutical drug products or medical devices are delineated in international pharmacopeial sterility test chapters such as USP <71> (30). There are two analytical choices: (a) passing the aqueous product through a membrane filter to collect the potential microorganisms recovered from its contents, followed by rinsing to remove residual antimicrobial preservative(s) contained in the product; or (b) adding the product directly into the enrichment media. The essential step before employing these validated compendial procedures is to run a product suitability test in the presence of a set of required QC microorganisms to ensure that, under the test conditions, the product ingredients do not interfere with the recovery and growth of these challenge QC microorganisms. The design of the suitability test establishes the procedures used during routine testing to provide scientific proof that the method will work under routine product-testing conditions.
对成品无菌药品或医疗器械进行的无菌检验采用的实验室方法在国际药典无菌检验章节中例如USP <71> (30)有描述。有两种分析选择:(a)将含水产品通过膜过滤器以收集从其内容物中复苏的潜在微生物,之后漂洗以去除产品中含有的残留抗菌防腐剂;或(b)将产品直接加入到浓缩介质中。采用这些经验证过的药典规程之前的关键步骤是在存在一系列所需的QC微生物情况下进行产品适用性试验,以确保在检验条件下,产品成分不会干扰这些挑战QC微生物的复苏和生长。适用性试验的设计建立了常规检验的规程,提供该方法在常规产品检验条件下有效的科学证据。
When the sterility test is properly performed as described in the compendia, and after determining the product suitability with that method, data integrity problems can result if sample handling and product testing is significantly altered or mismanaged from the approved SOPs. The following are risks for data integrity breaches in sterility testing:
当无菌检验按照药典中的描述正确运用,并确定该方法对产品的适用性后,如果样品处置和产品检验与已批准SOP相比被显著改变或管理不当可能会导致数据可靠性问题。以下是无菌检验中破坏数据可靠性的风险:
Before product containers (units) are transferred to the sterility testing suite or HEPA-filtered laminar flow hood, the outside of each container is usually subjected to a disinfection procedure: sprayed or wiped with a sporicidal solution to decontaminate the exterior of the container, preventing surface contamination from being transferred into the testing area. Any overexposure to the disinfection solution may penetrate the package or container and kill potential product contaminants before the sterility test is performed. This critical analytical step for certain product types (e.g., medical devices, combination products), if performed incorrectly, can result in a “false negative” sterility test that would allow the release of a potentially contaminated lot of parenteral or implantable products. This data integrity problem may not be obvious on a laboratory worksheet or detected in LIMS because the destruction of potential microbial results would have occurred before the product was tested.
In a pharmaceutical microbiology laboratory, the preparation of the media and reagents used during sterility testing is an important step, which should be performed carefully to avoid mistakes. For instance, when analyzing products by the “direct inoculation” approach, the accidental use of a lower volume of test medium may create an inhibitory growth condition due to a higher concentration of the product or preservatives in the test medium compared to those levels proven acceptable during the suitability test. The early detection of low volume test tubes can be missed when the tubes are located in the middle of the holding rack.
One observation that has been documented on several FDA Form 483s occurs when the membrane filter is transferred to the enrichment broth, or when a powdered product is added directly to the medium, and the membrane filter adheres to the top of the inner portion of the test tube above the liquid medium (Figure 5.2.4-1). This problem has been traced to the use of short, four-inch forceps rather than the recommended 10- or 12-inch forceps that may not allow the transfer of the membrane filter into the broth. A similar mishap may occur when hygroscopic powder products adhere to the neck of the inner tube, preventing the product from entering the enrichment media and allowing potential product contaminants to grow (Figure 5.2.4-2). A third example, documented during the inspections of contract laboratories performing sterility testing on sterile medical devices, involves ineffectively cutting up long catheters or complex devices, preventing the product from contacting the enrichment broth. The portions of the product protruding from the liquid broth may be the regions of the device that contain the indigenous microorganisms (Figure 5.2.4-3).
These examples reflect testing conditions that may have gone unrecorded or unobserved by a reviewing supervisor; however, their occurrence may result in false negative data to the final recorded laboratory result. The prevention of problems in the pharmaceutical microbiology laboratory that affect data integrity may need to be fixed at stages before observational or data entry is recorded on a worksheet or in LIMS.
这些例子反映了审查主管可能未记录或观察到的检验条件;然而,这些事件的发生会给最终记录的实验室结果的假阴性数据。防止制药微生物实验室影响数据可靠性的问题的发生,需要在观察或数据记录在工作表或LIMS之前处理。
5.2.5 Bacterial Endotoxin Testing 细菌内毒素检验
The bacterial endotoxin test (BET) described in compendial test chapters is an enzymatic assay that employs the reagent limulus amebocyte lysate (LAL) along with a vial of controlled standard endotoxin (CSE). The CSE serves as an artificial, laboratory-derived source of bacterial endotoxin that can be used to perform the suitability test (inhibition/enhancement test); it is also employed as a positive control to ensure that incubation conditions during product testing have not been compromised. The BET can also be conducted using a gel clot procedure or with the use of a spectrophotometer, employing a modified version of the LAL reagent (34). Each of these reagents requires careful handling and storage, the directions for which are clearly stated in the product's package instructions.
药典检验章节中描述的细菌内毒素检验是一种酶测定法,使用鲎阿米巴样细胞裂解物(LAL)以及内毒素标准品(CSE)试剂。CSE是一种人工的实验室衍生的细菌内毒素,可用于进行适用性试验(抑制/增强试验);也可用作阳性对照,以确保产品检验期间的孵育条件未受影响。BET也可以使用凝胶法,或改良式LAL试剂加分光光度计的方法(34)。这些试剂中的每一种都需要小心处理和储存,在产品的包装说明中有明确介绍。
To minimize potential data integrity problems with the final laboratory test results, when performing the BET method for finished product testing of pharmaceutical drugs or medical devices, there are a few critical parameters that need to be understood, satisfied, and documented:
为了最大程度地减少最终实验室检验结果的潜在数据可靠性问题,在对药品或医疗器械的成品检验执行BET方法时,需要了解,满足和记录一些关键参数:
During product container storage (refrigerated or at room temperature), when vial or ampoule samples containing aqueous solutions are delivered to the laboratory, the indigenous bacterial endotoxin may form micelles or attach to the glass or rubber stopper surfaces and move out of the solution. Unless the laboratory SOP for performing the BET on aqueous products includes a mixing step prior to removing the test aliquot, the detectable level of endotoxin determined by the assay will be underestimated and may inaccurately portray the product’s endotoxin unit value within specifications. Consequently, the final analytical results could be impacted, even when using a validated and properly controlled assay, depending on the outcome of a formal investigation (35).
识林解析:
尽管指南中提到的问题在1989年的文献(Guilfoyle, D.; Yager, J.; Carito, S. Effect of Refrigeration and Mising on Detection of Endotoxin in Parenteral Drugs Using the Limulus Amebocyte Lysate (LAL) Test. 1989, 43 (4), 183-187.))中已经有讨论,但识林警告信数据库中收录的12条内毒素相关的案例中,没有一个与低估内毒素水平相关。
An allowable amount of bacterial endotoxin (as defined in the pharmacopeial product monographs) may be present in a drug product based on its dosage, route of administration, and total amount of endotoxin delivered, e.g., within a one-hour dosing for an injectable product. A welldesigned BET method requires that the maximum valid dilution (MVD) be calculated before the assay is performed. If it is not calculated precisely, with the formula variables and the units used in the MVD formula, an incorrect value may result. The data integrity problem may occur if the calculated MVD value is higher than the true number. That would make it possible to run the assay at a higher product dilution, creating a testing situation where unacceptable levels of endotoxin may be present in the product but released without detection. Since endotoxin contamination can have dire patient consequences, this is a crucial data integrity matter.
In a U.S. Department of Justice criminal prosecution brought by the FDA involving a fraudulent and contaminated hormone product, the drug manufacturer submitted the bacterial endotoxin analytical data as part of the laboratory evidence. The FDA laboratory detected levels of bacterial endotoxin in the vials used for product rehydration above USP specifications. The drug manufacturer employed a private laboratory to test the same suspect lot for bacterial endotoxin and reported the levels of bacterial endotoxin detected as below the compendial specification. Laboratory records showed that the analyst for the private lab manipulated the product dilution to avoid detecting the actual level of endotoxin in the product. The analytical report had been tailored to give a finished result with an endotoxin level below the USP allowable limit. The FDA laboratory, however, performed the full range of product dilutions to avoid missing the actual endotoxin contamination level. The FDA’s test data proved the contract laboratory had manipulated the dilution to produce false results for their client. This example illustrates the need to both understand the analysis procedure and recognize how it can be manipulated.
1. U.S. FDA. Warning Letter No. 320-17-13 to Wockhardt, Ltd., dated 12/23/16.
2. U.S. FDA. Warning Letter No. 320-17-04 to Sekisui Medical Co., Ltd., dated 11/8/16.
3. U.S. FDA. Warning Letter No. 320-17-05 to Srikem Laboratories Pvt. Ltd., dated 11/8/16.
4. U.S. FDA. Warning Letter No. 320-17-03 to Beijing Taiyang Pharmaceutical Industry Co., Ltd., dated 10/19/16.
5. U.S. FDA. Warning Letter No. 320-17-02 to Interpharm Praha A.S., dated 10/18/16.
6. U.S. FDA. Warning Letter No. 320-16-07 to Ipca Laboratories Ltd. dated 1/29/16.
7. U.S. FDA. Warning Letter No. 320-15-17 to Unimark Remedies Ltd. dated 9/28/15.
8. U.S. FDA. Warning Letter No. 320-15-05 to Micro Labs Limited dated 1/9/15.
9. U.S. FDA. Warning Letter No. 320-15-01 to Sharp Global Limited dated 10/15/14.
10. U.S. FDA. Warning Letter No. 320-14-11 to Apotex Pharmachem India Pvt Ltd. dated 6/16/14.
11. U.S. FDA. Warning Letter No. 320-14-03 to Usv Limited dated 2/6/14.
12. U.S. FDA. 2015. Regulatory Action Against Ranbaxy.