首页
>
资讯
>
国内药政每周导读:Q1 中文版,临床试验风险管理,抗肿瘤 BE 和 PK 受试者,CDE 技术问答更新,参比目录公示93批
出自识林
国内药政每周导读:Q1 中文版,临床试验风险管理,抗肿瘤 BE 和 PK 受试者,CDE 技术问答更新,参比目录公示93批
2025-05-12
【早期研发与临床】
5.6,【CDE】关于公开征求《创新药研发期间风险管理计划撰写技术指导原则(征求意见稿)》意见的通知
征求意见截至6月6日。
区别于上市后风险管理计划(RMP),创新药研发期间风险管理计划(Development Risk Management Plan,DRMP)是临床试验 期间风险管理 的重要工具。监管机构可通过该文件,快速获知申请人对临床试验的风险识别及控制措施,评估控制措施是否充分。申请人可通过该文件的起草和不断更新,及时评估临床试验风险,采取必要的控制措施,严格落实药物安全主体责任。
另外,为保持与上市后风险管理计划(Risk Management Plan,RMP)的衔接性,参考CIOMS VI,统一上市前文件名称为DRMP。
本指导原则共五章,详细介绍了创新药DRMP撰写原则、一般考虑和主要内容,并提供了模板供撰写时参考。
5.7,【CDE】关于公开征求《抗肿瘤药物生物等效性及药代动力学比对研究受试者人群选择考虑(征求意见稿)》意见的通知
征求意见截至6月7日。
抗肿瘤药物 的特殊性决定了其生物等效性 及药代动力学 比对研究(以下简称 BE/PK 比对研究 )中受试者的选择不同于一般非抗肿瘤药物,除科学性因素外,还需格外关注受试者 的安全。不同类型抗肿瘤药物安全性风险不同,对于受试者的选择考虑因素亦不同。
本指导原则主要基于小分子化学药物及单抗 类药物的研究经验,为抗肿瘤药物化学仿制药 生物等效性研究与生物类似药 药代动力学比对研究中受试者人群的选择考虑提供建议。
抗肿瘤药物 BE/PK 比对研究受试者人群的选择,需结合产品的作用机制、已有非临床、临床信息等,采用证据权重方法评估受试者人群的适用性(见第三章)。经评估可选择健康受试者时,建议关注药物的生殖毒性 、遗传毒性、对受试者生长发育的影响等;选择目标适应症 患者时,需关注受试者的获益、试验过程中可能对对试验结果产生影响的因素等(见第四章)。申请人、机构伦理审查委员会、研究者均应从各自责任出发保障受试者权益(见第五章)。
【注册与变更】
5.9,【CDE】常见一般性技术问题解答 - 新增2个问答
新增问题是:
对于化学药物活性成份A与化学药物活性成份B形成的共晶 制剂,该如何注册申报?
截至2025-05-09,官网共发布一般性技术问题解答 245条(其中有25条重复,实际共计220个)。会员还可点击CDE 化学仿制药共性问题 、CDE 受理共性问题 查看其他问题清单。
【CMC药学研究】
5.6,【CDE】关于公开征求 ICH《Q1:原料药和制剂的稳定性试验》指导原则草案意见的通知
征求意见截至7月15日。
4月18日,ICH发布了《Q1:原料药和制剂的稳定性试验》第二阶段指南文件 ,正式向公众征求意见。该指南整合现行7份指南(包括:ICH Q1A -F 系列和Q5C ,并移除了以前指南标题里醒目的New字样)的全部内容,将其合并为一个全面的稳定性指南 ,目标是简化指南体系,促进对核心稳定性 原则的统一解释,填补潜在的空白和模糊领域,同时引入新的技术议题,如风险管理 的创新工具和先进疗法 的稳定性考虑。
修订历程始于2022年11月,当时成立了专家工作组(Expert Working Group,简称EWG),批准了一份指南修订概念性文件 。随后,经过多次草案的发布、反馈和修订,最终在2025年4月形成了这一步文件。该文件将在公众意见征询后进一步修订,并在后续步骤中完善。按概念性文件中的规划,正式指南(即第四阶段文件)将于2025年第4季度落地。
更多内容请点击《ICH 发布新 Q1 稳定性指南征求意见,整合7份指南》 。
识林会员可点击“中国ICH指南官方翻译和适用公告 ”查阅国内全部ICH指南内容。
5.7,【CDE】关于公开征求《化学仿制药参比制剂目录(第九十三批)》(征求意见稿)意见的通知
截至目前,NMPA已发布参比制剂目录91批,CDE已公示93批。
识林用户可登录“中国参比制剂库 ”查阅。
【新药批准和报产】
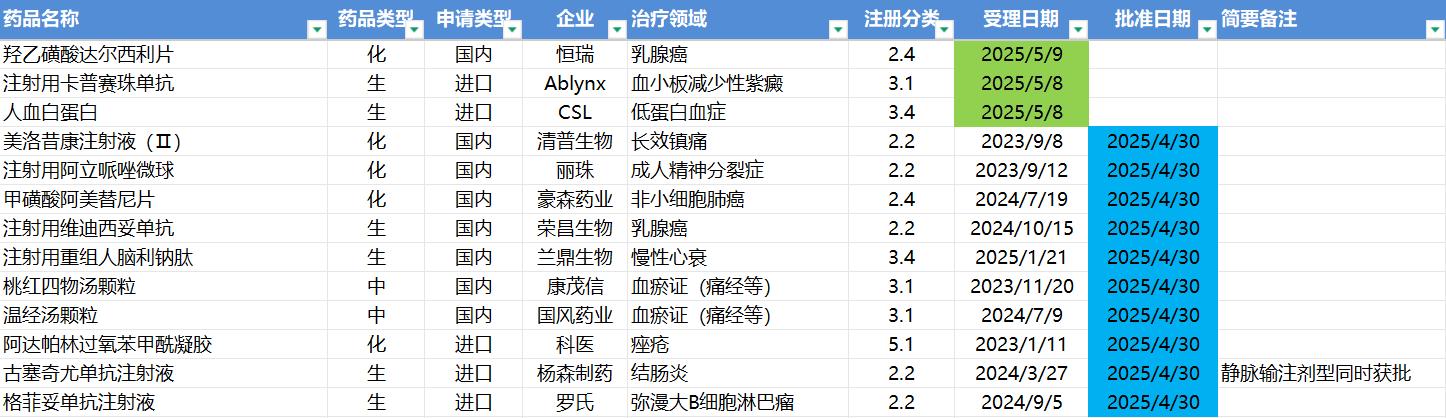
5.5-5.11,NMPA发布10个新药批准,CDE受理3个NDA
注:仅列出新药(包括改良型新药 )的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症 、临床研究策略、注册路径,都值得业界关注和分析。
识林® 版权所有,未经许可不得转载
适用岗位及工作建议:
注册(RA) :必读。需关注参比制剂目录更新,及时调整注册策略,确保申报资料与最新要求一致。研发(R&D) :必读。根据参比制剂目录调整研发计划,确保产品质量与参比制剂一致。质量管理(QA) :必读。监督生产过程,确保产品质量符合参比制剂标准。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
注册 :必读。需对照文件更新申报资料,重点关注元素杂质风险评估报告、溶出曲线对比数据及变更分类依据。 研发 :必读。在处方设计、工艺开发中严格遵循参比制剂标准,强化溶出度方法学验证及稳定性试验设计。 QA/QC :必读。完善元素杂质检测方法(如ICP-MS),制定原料药和制剂的质量控制策略,确保数据符合ICH Q3D要求。 生产 :必读。针对批量变更、辅料替换等需执行工艺验证,并保留批生产记录备查。 以上仅为部分要点,请阅读原文,深入理解监管要求。
以下为对CDE受理共性问题文件的概要分析:
【文件概要】
【适用范围】
【影响评估】
【实施建议】
注册 :必读。需对照更新申报策略,尤其关注多肽类制剂、变更豁免等特殊情形分类规则。 CMC :必读。严格遵循电子申报资料结构(如模块文件夹命名)、签章有效性校验及光盘刻录标准。 临床 :关注临床试验期间剂型变更需重新申报的要求,避免研发中断。 RA :必读。主导跨部门协调,确保电子申报全流程(如申请表签章、资料完整性)符合新规。 以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
注册 :必读。需对照文件调整申报策略,如多肽类制剂按境内外上市情况选择分类,境外变更文件豁免需符合指南条件。 QA :必读。关注非无菌制剂微生物控制标准更新,确保生产工艺与注册标准一致。 研发 :必读。罕见病药物研发需评估是否符合临床试验减免条件,共晶制剂需明确活性成分分类依据。 临床 :必读。临床试验期间剂型变更需重新申请,早期试验适应症填写需备注多队列研究细节。 生产 :必读。批生产记录需包含关键批次,电子申报资料需严格遵循光盘刻录及签章规范。 以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保质量保证团队理解稳定性指南的更新,以符合核心稳定性原则。 研发:研发团队需关注新技术分析和现代工具/策略,以增强产品理解。 注册:注册团队应了解更新后的指南,以确保注册文件符合最新的国际协调标准。 临床:临床团队需了解稳定性数据如何影响临床试验设计和药品供应。 文件适用范围:
文件要点总结:
指南整合与简化 :将ICH Q1A-F和Q5C指南整合为单一指南,专注于核心稳定性原则,简化系列指南。解释一致性 :通过重组为具有特定主题附件/附录的核心指南,明确指南各部分适用于哪些产品类型,提高指南解释的一致性。技术组件澄清 :包括对活性药物成分、中间体、药品的稳定性原则的共同点和重叠点的合并,以及特定产品类型的稳定性概念。新技术与工具 :指南将包含对增强产品理解的现代分析技术和工具的指导,如稳定性建模和风险管理。质量体系相关主题 :涉及产品供应链、生产过程中的稳定性考虑,以及OOS和OOT稳定性数据的处理。以上仅为部分要点,请阅读原文,深入理解监管要求。
关键日期:本文件为 ICH Q1A(R2) 指南的当前步骤 4 版本,日期为 2003 年 2 月 6 日。该指南已被 ICH 专家工作组制定,并已根据 ICH 流程由监管方咨询。本指南在 ICH 流程的第 4 步被推荐采纳。
适用范围:本文件适用于新药物物质和产品的稳定性测试,包括化学药品、生物制品、疫苗和中药等。适用于注册分类(创新药或仿制药、生物类似药、原料药等)和监管市场(如中国、美国、欧盟等)。适用于 Biotech、大型药企、跨国药企、CRO 和 CDMO 等企业类型。
适用岗位:本文件对药品研发、注册、质量保证(QA)、生产、市场等岗位的工作带来变化,特别是对稳定性研究和注册申报的岗位是必读。
文件要点总结:
稳定性测试目的:为证明药物物质或药物产品在各种环境因素(如温度、湿度、光照)影响下随时间变化的质量,并建立药物物质的复验期或药物产品的货架期及推荐储存条件。 稳定性测试条件:基于对 EC、日本和美国三个地区气候条件影响的分析,世界被划分为四个气候区域 I-IV,本指南针对气候区域 I 和 II。 药物物质稳定性测试:包括压力测试、批次选择、容器封闭系统、规格、测试频率、储存条件、稳定性承诺、评估和标签声明等。 药物产品稳定性测试:包括光稳定性测试、批次选择、容器封闭系统、规格、测试频率、储存条件、稳定性承诺、评估和标签声明等。 稳定性数据包:本指南定义了新药物物质或药物产品注册申请所需的核心稳定性数据包,同时允许足够的灵活性以适应由于特定科学考虑和被评估材料特性可能出现的不同实际情况。 以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):负责确保稳定性测试符合指南要求,并监督实施。 注册(Regulatory Affairs):需要理解指南内容,以确保注册文件和申报材料符合规定。 研发(R&D):在药物研发阶段,依据指南进行稳定性研究设计和数据分析。 生产(Production):根据指南要求进行产品的稳定性测试和生产管理。 工作建议:
QA:监控稳定性测试流程,确保所有操作符合ICH Q1指南的要求,及时更新SOP以符合最新指南。 注册:在准备注册文件时,确保包含所有必要的稳定性数据,并根据指南要求解释数据。 研发:设计实验时考虑指南中提到的所有关键质量属性,确保研究结果能够支持产品的再测试期或货架寿命。 生产:在生产过程中,遵循指南中的存储条件要求,确保产品质量和稳定性。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
法规指南解读:ICH_Q5C_Stability_Testing_of_Biotechnological/Biological_Products
适用岗位(必读):
QA(质量保证):确保稳定性测试程序符合ICH Q5C指南要求。 注册:在药品注册文件中准确反映稳定性数据和条件。 研发:设计符合ICH Q5C要求的生物技术/生物制品稳定性研究方案。 生产:根据指南要求进行生物制品的生产和储存。 工作建议:
QA应定期审核稳定性测试程序,确保符合ICH Q5C的最新要求。 注册人员需在注册文件中明确指出稳定性测试条件和结果。 研发团队应根据指南制定详细的稳定性测试方案,并确保测试方法的验证。 生产部门需确保生产规模的批次符合ICH Q5C的稳定性数据要求。 文件适用范围:
文件要点总结:
特殊特性考虑 :生物技术/生物制品的稳定性测试需考虑分子构象和生物活性的维持,对环境因素特别敏感。稳定性测试程序 :应包括适当的分析方法,确保产品在拟定储存期内的稳定性。批次选择 :至少3批代表性的原料药或最终产品,用于长期稳定性研究。稳定性指示特性 :开发包括效力、纯度和分子特性的稳定性指示特性,确保检测产品变化。储存条件 :精确定义储存温度和条件,考虑加速和压力条件下的稳定性。以上仅为部分要点,请阅读原文,深入理解监管要求。