首页
>
资讯
>
ICH 发布新 Q1 稳定性指南征求意见,整合7份指南
出自识林
ICH 发布新 Q1 稳定性指南征求意见,整合7份指南
2025-04-22
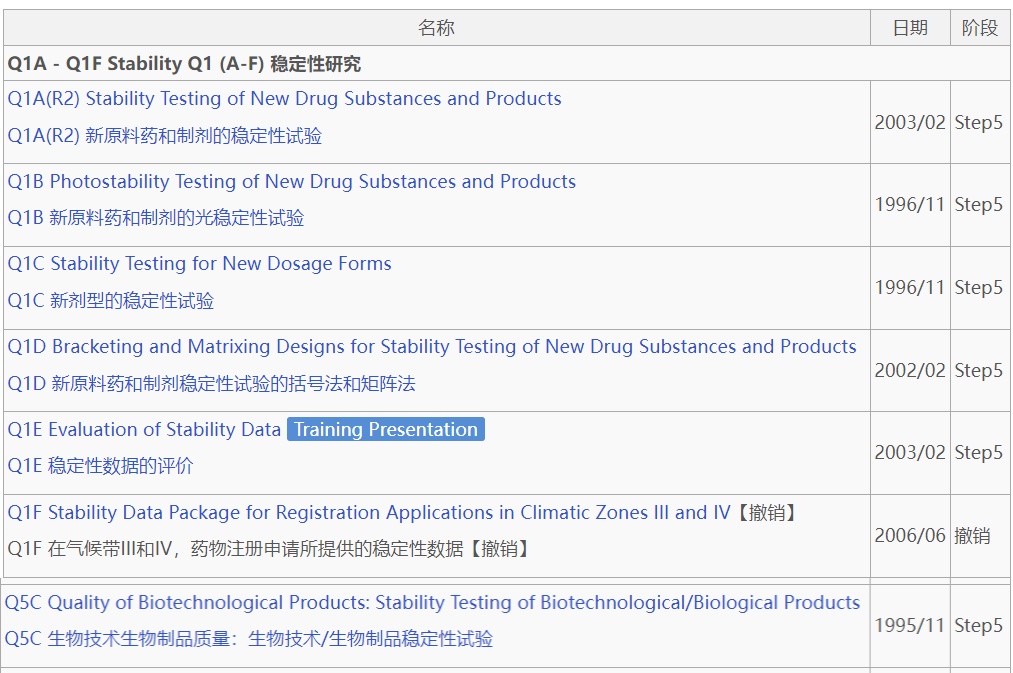
2025年4月18日,ICH发布了《Q1:原料药和制剂的稳定性试验》第二阶段指南文件 ,正式向公众征求意见。该指南整合现行7份指南(包括:ICH Q1A -F 系列和Q5C ,并移除了以前指南标题里醒目的New字样)的全部内容,将其合并为一个全面的稳定性指南 ,目标是简化指南体系,促进对核心稳定性 原则的统一解释,填补潜在的空白和模糊领域,同时引入新的技术议题,如风险管理 的创新工具和先进疗法 的稳定性考虑。
修订历程始于2022年11月,当时成立了专家工作组(Expert Working Group,简称EWG),批准了一份指南修订概念性文件 。随后,经过多次草案的发布、反馈和修订,最终在2025年4月形成了这一步文件。该文件将在公众意见征询后进一步修订,并在后续步骤中完善。按概念性文件中的规划,正式指南(即第四阶段文件)将于2025年第4季度落地。
ICH建议药企在审查草案指南时,应先通读整个指南,因为其组织结构与现有ICH稳定性指南不同,某些内容可能在不同章节中呈现。同时ICH强调指南无法提供所有产品特定场景的指导。此外,指南允许在科学合理的情况下采用替代方法。
整合7份指南的新架构
新版ICH Q1征求意见稿包含18个章节,涵盖了从稳定性研究的设计到数据评估、标签声明以及产品生命周期 管理的各个方面。此外,还包括3个附录,分别提供关于稳定性方案设计、稳定性建模和先进疗法药品(Advanced Therapy Medicinal Products,简称ATMPs)稳定性的补充指导。
各章节主要内容简介如下:
1. 引言(Introduction)
引言章节为理解指南的范围和与现有稳定性指南的格式差异提供了基础。ICH建议读者首先阅读此章节,并在阅读指南时参考。
2. 强力和强制条件下的开发稳定性研究(Development Stability Studies Under Stress and Forced Conditions)
该章节指导如何利用早期开发阶段生成的数据(如了解内在稳定性、潜在降解产物 /途径、确认方法适用性以及支持方法验证 )来设计稳定性方案并支持长期储存。此章节内容整合了现有ICH Q1A 和/或Q5C 中关于强力稳定性试验和强制降解的内容。
3 - 7. 长期稳定性方案设计(Protocol Design for Formal Stability Studies)
这些章节旨在共同建立长期稳定性方案。它们结合、对齐、澄清并升级了ICH Q1A和Q5C中关于主要稳定性研究的内容,提供了关于主要稳定性方案的具体指导、提交时的最低数据建议、属性选择以及代表性批次的识别,明确了长期和加速储存条件,并阐述了这些原则如何适用于其他方案,如稳定性承诺或支持批准后变更 的方案。
8 - 11. 补充稳定性研究(Supplementary Stability Studies)
这些章节提供了补充主要稳定性研究的其他研究指导。
光稳定性 (Photostability):与ICH Q1B 一致,为药品的推荐储存条件提供信息。此章节指导应与第2节的指导结合使用。
中间体的加工和储存时间稳定性(Stability Considerations for Processing and Holding Times for Intermediates):新引入内容 ,明确了何时应在监管提交中包含数据,何时可在药品质量体系(Pharmaceutical Quality System,简称PQS)内管理数据。
短期储存条件(Short-Term Storage Conditions):新引入内容 ,为支持标签上包含短期储存条件的产品提供指导。此章节不适用于所有药品。
使用中稳定性(In-Use Stability):新引入内容 ,为支持药品的使用期限和储存条件提供指导。
12. 标准物质 、新型辅料 和佐剂(Reference Materials, Novel Excipients and Adjuvants)
新引入内容 ,提供了如何使用ICH Q1草案指南的其他章节关于标准物质、新型辅料和佐剂的指导。
13. 数据评估(Data Evaluation)
该章节提供了数据评估的指导,包括重新检验期和有效期的一般考虑、数据的统计评估、外推和呈现。内容与现行的ICH Q1E 一致,并应在应用统计方法时与附录2结合使用。
14. 标签 (Labelling)
该章节提供了关于标签和储存声明以及从稳定性 研究中得出的超出标签声明的偏离的指导。
15. 稳定性承诺和产品生命周期 管理(Stability Considerations for Commitments and Product Lifecycle Management)
该章节提供了关于产品生命周期管理的稳定性承诺的指导,包括批准后变更 的新指导。关于承诺的内容与ICH Q1A 一致,关于引入新剂型 的内容与ICH Q1C 一致。
16 - 17. 术语表和参考文献(Glossary and References)
这些章节提供了术语表和参考文献。
18. 附录(Annexes)
指南草案 包括3个附录:
附录1(Annex 1):包括关于括号法 和矩阵法 的指导,目前包含在ICH Q1D 中。提供了基于知识和风险的方案减少的新指导。
附录2(Annex 2):提供稳定性建模的指导。包括目前在ICH Q1E 中提供的指导以及使用增强稳定性建模的新指导。
附录3(Annex 3):新引入内容 ,提供关于ATMPs稳定性的指导。该附录是核心ICH Q1草案指南的补充,而不是ATMPs 的独立指南。
识林-实木
识林® 版权所有,未经许可不得转载
岗位必读建议:
QA:确保质量保证团队理解稳定性指南的更新,以符合核心稳定性原则。 研发:研发团队需关注新技术分析和现代工具/策略,以增强产品理解。 注册:注册团队应了解更新后的指南,以确保注册文件符合最新的国际协调标准。 临床:临床团队需了解稳定性数据如何影响临床试验设计和药品供应。 文件适用范围:
文件要点总结:
指南整合与简化 :将ICH Q1A-F和Q5C指南整合为单一指南,专注于核心稳定性原则,简化系列指南。解释一致性 :通过重组为具有特定主题附件/附录的核心指南,明确指南各部分适用于哪些产品类型,提高指南解释的一致性。技术组件澄清 :包括对活性药物成分、中间体、药品的稳定性原则的共同点和重叠点的合并,以及特定产品类型的稳定性概念。新技术与工具 :指南将包含对增强产品理解的现代分析技术和工具的指导,如稳定性建模和风险管理。质量体系相关主题 :涉及产品供应链、生产过程中的稳定性考虑,以及OOS和OOT稳定性数据的处理。以上仅为部分要点,请阅读原文,深入理解监管要求。
关键日期:本文件为 ICH Q1A(R2) 指南的当前步骤 4 版本,日期为 2003 年 2 月 6 日。该指南已被 ICH 专家工作组制定,并已根据 ICH 流程由监管方咨询。本指南在 ICH 流程的第 4 步被推荐采纳。
适用范围:本文件适用于新药物物质和产品的稳定性测试,包括化学药品、生物制品、疫苗和中药等。适用于注册分类(创新药或仿制药、生物类似药、原料药等)和监管市场(如中国、美国、欧盟等)。适用于 Biotech、大型药企、跨国药企、CRO 和 CDMO 等企业类型。
适用岗位:本文件对药品研发、注册、质量保证(QA)、生产、市场等岗位的工作带来变化,特别是对稳定性研究和注册申报的岗位是必读。
文件要点总结:
稳定性测试目的:为证明药物物质或药物产品在各种环境因素(如温度、湿度、光照)影响下随时间变化的质量,并建立药物物质的复验期或药物产品的货架期及推荐储存条件。 稳定性测试条件:基于对 EC、日本和美国三个地区气候条件影响的分析,世界被划分为四个气候区域 I-IV,本指南针对气候区域 I 和 II。 药物物质稳定性测试:包括压力测试、批次选择、容器封闭系统、规格、测试频率、储存条件、稳定性承诺、评估和标签声明等。 药物产品稳定性测试:包括光稳定性测试、批次选择、容器封闭系统、规格、测试频率、储存条件、稳定性承诺、评估和标签声明等。 稳定性数据包:本指南定义了新药物物质或药物产品注册申请所需的核心稳定性数据包,同时允许足够的灵活性以适应由于特定科学考虑和被评估材料特性可能出现的不同实际情况。 以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读指南
QA:负责确保产品质量符合光稳定性测试要求。 研发:进行新药物质和产品的光稳定性测试,以评估光对药物的影响。 注册:在药品注册申请中包含光稳定性测试信息。 文件适用范围
本文适用于新分子实体及其相关药物产品的光稳定性测试,包括化学药品和生物制品。适用于欧盟、日本和美国监管机构,主要针对大型药企和Biotech公司。
文件要点总结
光稳定性测试重要性 :强调光测试是压力测试的一部分,用于评估药物在光照条件下的稳定性。样品呈现 :建议对样品进行适当的物理状态考虑,以最小化光照测试中可能发生的物理变化。分析方法 :样品在光照暴露后应使用适合的分析方法检测物理性质变化和降解产物。结果判断 :通过强制降解研究和确认性研究,确定是否需要特殊的制造、配方或包装措施,以及是否需要光阻隔包装或特殊标签。测试条件 :样品应暴露在特定的光照条件下,以确保获得足够的光暴露量,包括总光照量和近紫外线能量。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:确保稳定性测试设计符合ICH Q1D指南要求,为减少设计提供合理性论证。 研发(R&D):在设计新药物质和产品的稳定性研究时,考虑应用括号法和矩阵法设计。 注册(Regulatory Affairs):在药品注册文件中准确体现稳定性研究设计,确保符合监管要求。 文件适用范围:
文件要点总结:
减少设计适用性 :提供了减少设计(括号法和矩阵法)的适用情况和前提条件,强调了在多因素设计中减少设计的合理性。括号法设计 :详细说明了括号法设计的因素、考虑因素和潜在风险,以及如何应用括号法设计。矩阵法设计 :介绍了矩阵法设计的因素、设计考虑、示例、适用性和减少程度,以及潜在风险。数据评估 :强调了减少设计研究中稳定性数据的评估应与完整设计研究相同。设计变更 :如果在研究过程中需要从减少设计变更为完整设计,应提供合理性论证,并根据完整设计和减少设计的原则进行适当调整。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位:
工作建议:
QA:确保稳定性数据的评估和呈现符合ICH Q1E指南的要求,监督稳定性研究的执行和数据分析。 注册:在注册文件中准确引用ICH Q1E指南,确保提交的数据和分析符合监管机构的要求。 研发:在药物开发过程中,应用ICH Q1E指南的原则来设计和执行稳定性研究。 生产:了解ICH Q1E指南中关于稳定性数据评估的要求,以确保生产过程中的控制和产品质量。 适用范围:
文件要点总结:
稳定性数据评估: 强调了使用ICH Q1A(R)指南中详述的原则生成的稳定性数据来提出注册申请中的复验期或货架期。统计分析: 明确了在评估稳定性数据时,应采用统计分析方法,包括线性回归和批次数据池化测试。数据呈现: 规定了所有属性的数据应以适当格式呈现,并包括对这些数据的评估。外推法: 详细描述了在长期数据基础上,如何合理使用外推法来延长复验期或货架期。不同存储条件下的数据评估: 提供了针对室温、冷藏和冷冻条件下储存的药物物质或产品的稳定性数据评估指南。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):负责确保稳定性测试符合指南要求,并监督实施。 注册(Regulatory Affairs):需要理解指南内容,以确保注册文件和申报材料符合规定。 研发(R&D):在药物研发阶段,依据指南进行稳定性研究设计和数据分析。 生产(Production):根据指南要求进行产品的稳定性测试和生产管理。 工作建议:
QA:监控稳定性测试流程,确保所有操作符合ICH Q1指南的要求,及时更新SOP以符合最新指南。 注册:在准备注册文件时,确保包含所有必要的稳定性数据,并根据指南要求解释数据。 研发:设计实验时考虑指南中提到的所有关键质量属性,确保研究结果能够支持产品的再测试期或货架寿命。 生产:在生产过程中,遵循指南中的存储条件要求,确保产品质量和稳定性。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
法规指南解读:ICH_Q5C_Stability_Testing_of_Biotechnological/Biological_Products
适用岗位(必读):
QA(质量保证):确保稳定性测试程序符合ICH Q5C指南要求。 注册:在药品注册文件中准确反映稳定性数据和条件。 研发:设计符合ICH Q5C要求的生物技术/生物制品稳定性研究方案。 生产:根据指南要求进行生物制品的生产和储存。 工作建议:
QA应定期审核稳定性测试程序,确保符合ICH Q5C的最新要求。 注册人员需在注册文件中明确指出稳定性测试条件和结果。 研发团队应根据指南制定详细的稳定性测试方案,并确保测试方法的验证。 生产部门需确保生产规模的批次符合ICH Q5C的稳定性数据要求。 文件适用范围:
文件要点总结:
特殊特性考虑 :生物技术/生物制品的稳定性测试需考虑分子构象和生物活性的维持,对环境因素特别敏感。稳定性测试程序 :应包括适当的分析方法,确保产品在拟定储存期内的稳定性。批次选择 :至少3批代表性的原料药或最终产品,用于长期稳定性研究。稳定性指示特性 :开发包括效力、纯度和分子特性的稳定性指示特性,确保检测产品变化。储存条件 :精确定义储存温度和条件,考虑加速和压力条件下的稳定性。以上仅为部分要点,请阅读原文,深入理解监管要求。