|
首页
>
资讯
>
FDA 分析过去七年 GCP 检查,揭示临床试验合规现状
出自识林
FDA 分析过去七年 GCP 检查,揭示临床试验合规现状
2025-08-13
FDA研究人员在7月份发表了一项针对2017至2023年间GCP检查的深度分析结果,显示合规率稳步提升,试验方案依从性成首要缺陷。
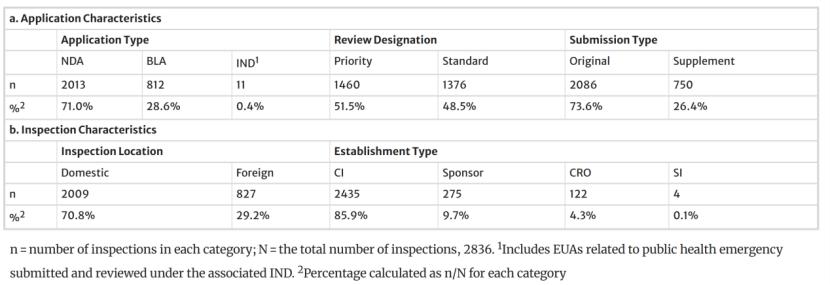
研究聚焦于支持新药申请(NDA)、生物制品许可申请(BLA)及紧急使用授权(EUA)的常规GCP检查,共涵盖2836次对临床研究者(CI,85.9%)、申办方(9.7%)、合同研究组织(CRO,4.3%)及申办方-研究者(Sponsor-Investigators,SI,0.1%,指的是既是发起者也是实施者的个人研究者)的检查,在该研究中大多数GCP检查针对的是原始审评(73.6%),优先审评和标准审评的检查数量大致相当,70.8%为美国境内检查,29.2%为境外检查。
检查结果显示整体合规性持续向好
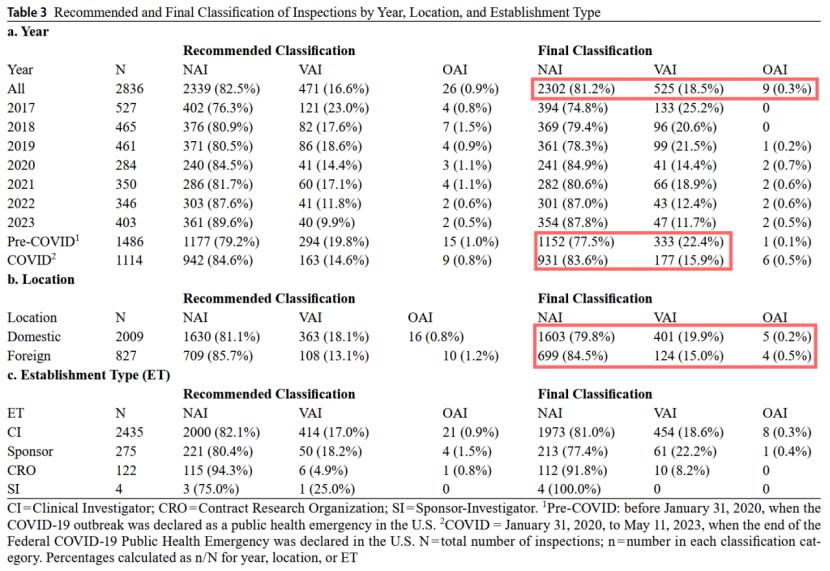
99.7%的检查最终分类为无行动指示(NAI,81.2%)或需自愿行动指示(VAI,18.5%),仅0.3%官方行动指示(OAI)。其中,境外检查的NAI比例(84.5%)略高于境内(79.8%),而境内VAI(19.9%)的比例略高于境外(15.0%),OAI的比例两者几乎相当(境外0.5%,境内0.2%)。
此外,FDA 483表格的签发率显著下降,从2017年的23.5%降至2023年的10.4%,这反映出行业GCP实践不断优化。同时,约97%的NAI建议(recommended)和96%的VAI建议最终(final)维持原分类,这意味着检查部门OSI的检查结果大部分得到了评估部门OII最终确认,判定高度一致。
可以看到,COVID-19疫情对检查进程产生了显著影响。2020年检查量骤降至284次,且2020–2022年间,VAI建议降级为NAI(降级意味着FDA最终判定某缺陷项的风险较低,反之则较高)的比例显著上升,FDA认为这一异常趋势与疫情期间“不可避免的方案偏离”有关。此外,在疫情期针对优先审评申请的检查比例反超标准审评,体现资源倾斜策略(见下图红框)。
试验方案偏离与记录问题最常见
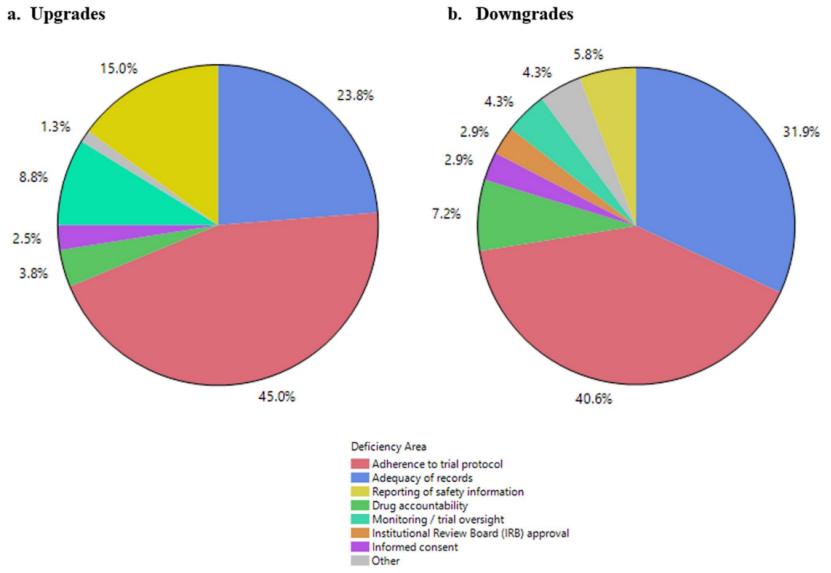
在60项升级检查(如NAI→VAI)和39项降级检查(如VAI→NAI)中,试验方案依从性缺陷占比最高(占升级案例的45%、降级案例的40.6%,可以理解为方案依从性风险的判定有高有低,但仍然最常见),记录充分性问题次之(升级占23.8%、降级占31.9%),其他缺陷包括安全性信息报告缺陷(升级占15%)、药物清点(drug accountability,指对临床试验用药品的接收、储存、分发、使用、归还和销毁进行完整记录和追踪的管理过程,以确保每一份药品的流向清晰可查)、监查/试验监督、机构审查委员会(IRB)批准、知情同意等。
研究指出,推广电子化系统(eSystems)可提升合规性,但方案依从性与记录规范性仍是全球临床合规的一大问题。文献还提及疫情期间,FDA通过远程监管评估(RRA)替代现场检查,并优先审查关键申请,确保工作连续性。技术进步与监管灵活性推动了合规提升,但基础操作仍需倍加关注。
自2015年“722”后,十年创新药发展,我国临床合规有了显著进步,也已获得国际认可,其标志是多个创新药在美国上市。从该文献笔者想到识林报道FDA宣布扩大对海外食品、药品生产设施的突击检查范围,取消提前通知,以实现国内外监管标准一致。此举目的在于解决“双重标准”的问题,因为突击检查能更真实地发现问题(国外缺陷率是国内两倍)。尽管研究显示FDA以前的GCP检查似乎并未针对境外场地,但未来趋势仍不可不察。
该研究有利于我国药企、临床中心以及CRO理解FDA检查实践,并为国内的申办者/研究者提供GCP合规洞察,值得细读。
识林-梓
识林®版权所有,未经许可不得转载
岗位必读建议: - 研发(R&D):应深入理解IND的提交要求、临床试验阶段及相应变更流程,确保研发过程中符合FDA规定。
- 临床(Clinical):必须熟悉临床试验的各个阶段,包括患者入组、数据收集和安全报告等,以保障试验顺利进行。
- 注册(Regulatory Affairs):需掌握IND的提交流程、年度报告和安全报告的要求,以确保合规提交。
- 质量管理(QA):应监督整个IND流程,确保所有记录和报告符合FDA规定,并在必要时进行审计。
文件适用范围: 本文适用于在美国进行的临床研究,涉及化学药、生物制品等药品类型,包括创新药、仿制药、生物类似药等注册分类。适用于Biotech、大型药企、跨国药企等企业类别。由美国食品药品监督管理局(FDA)发布。 文件要点总结: - IND申请要求:明确了提交IND的必要条件,包括药物的临床试验阶段和IND内容格式。
- 临床试验阶段:将临床试验分为三个阶段,每个阶段都有其特定目的和要求。
- IND内容和格式:详细列出了IND应包含的内容,如药物信息、临床试验方案、药物的化学制造与控制信息等。
- 安全报告:强调了对不良事件的监测和报告流程,确保患者安全。
- IND的变更和年报:规定了对IND进行变更的流程和年度报告的要求。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |