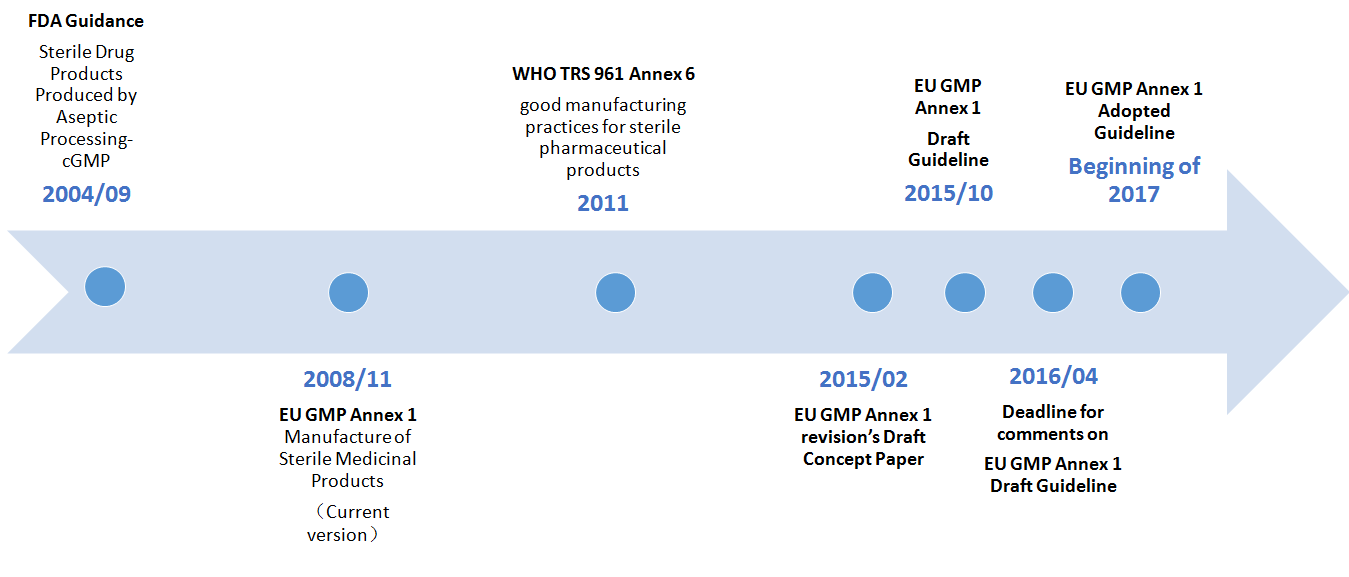
Comments on the Annex 1 revision's concept paper are due March 31. Also, there will be another six months to comment after EMA and PIC/S issue a draft guideline in October. EMA and PIC/S previously revised the Annex 1 guideline in 1996, 2003 and 2007, but none of those revisions were as thorough as the one they' reconducting now.Why this revision? The guideline needs to reflect recent advances in technology as well as the International Conference on Harmonization's Q9 risk management and Q10 quality systems guidelines. There also are some historical inaccuracies and ambiguities in the document that the authorities intend to address.
Hopkins, an inspector with the United Kingdom's Medicines Healthcare products Regulatory Agency, added some reasons of his own. The industry is losing its technical skill level. Much work has shifted to emerging economies where manufacturers still are “learning by deficiencies.”
Also, there are new and emerging technologies, and areas still not covered by the guideline, such as media fills, small batches, active pharmaceutical ingredient campaigns, closed systems and blow fill seal technologies.
PDA established an expert group that began meeting in July and on March 13 issued Part 1 of a new two-part points-to-consider document. “Rather than just present what people are doing in the industry or what our position might be in regard to what regulators are asking for, what we decided to do was focus on real science,” PDA Chair Hal Baseman told reporters March 16 at the PDA annual meeting in Las Vegas.
One attendee speaking in a later session that day attributed this historical standard to the fan speed that happened to be available to the World War II Navy engineer who invented the HEPA filter.
“What is the state of the art there?” Baseman asked. “Well, the state of the art is not a number. The state of the art is to go in and look at your clean room operation, take a combination of dynamic air profile, smoke studies if you will, particulate levels, what's going on there, and devise from that what is the optimal velocity. … Today, the state of the art is to take a true risk-based design approach to it.”
Gabriele Gori, GSK Vaccines, who with Baseman co-chairs PDA's points-to-consider task force, on March 18 described key positions PDA is taking in Part 1 of its points-to-consider document and requested input on possible additional topics for Part 2. Gori also updated the PDA meeting on the work of another task force, co-chaired by Jette Christensen, Novo Nordisk, and Bob Darius, GSK, that is comparing three sterile manufacturing GMP guidelines –the 2004 FDA guidance,a 2011 World Health Organization technical report annex and the current 2008 version of the EU GMP Annex 1. PDA plans to publish that workgroup's detailed comparison in May with the idea of encouraging regulators to identify key differences between the documents and perhaps further align their expectations
PDA has so far developed 70 points consider, 38 of which it addressed in Part 1 of the document. Gori described some of the key positions in his remarks.
Airflow Velocity Measurements
Unlike other GMP guides, Annex 1 gives a value for airflow velocity at the working position rather than at the HEPA filter face. PDA accepts the value, 0.45 meters per second, plus or minus 20%, but wants it measured 15 to 30 centimeters from the filter face. The association would add that the airflow should be unidirectional and sufficient to protect exposed product as well as packaging components and surfaces that contact the product.
Grade A Environment Over Cappers
There is no need for Annex 1 to continue requiring Grade A air over capping stations outside the aseptic core, or, as PIC/S clarified in 2010, near the HEPA filters that supply air to the capping stations, PDA says.
Any vial that is properly stoppered in the aseptic core is adequately sealed against microbes while en route to the capping station, and can be capped under air that is merely HEPA filtered, PDA says.
In PDA's view, the key issue, Gori explained, is that “you have to have a sound automatic system to ensure that you are rejecting any vial which is not properly sealed by the stopper before it is crimped.”
Laminar vs. Unidirectional Airflow
PDA says Annex 1 should require unidirectional rather than laminar airflow in Grade A critical zones because laminar airflow is unnecessary and virtually impossible.
The key, the association said, is to establish unidirectional flow such that exposed product and sterile components always receive “first air” that hasn't passed any other component, operator or equipment that hasn't been sterilized. PDA adds that it believes turbulent airflow may be OK in closed isolators.
≥0.5μm and ≥5μm Total Particle Monitoring
Over the years, industry has chafed at the Annex 1 requirement to apply limits to air particles in Grade A environments and at-rest Grade B environments that are five microns or more in size.
Pointing to a draft ISO standard that says such particles are too few in such environments and too large for reliable sampling, PDA recommends merely trending such particle counts rather than setting a limit to them.
PDA agrees with the overlapping requirement for limits on particles that are 0.5 microns or larger.
PDA recommends that manufacturers be expected to choose temperature regimens for environmental sample incubation that are appropriate for the microorganisms of concern, and suggests that the use of non-selective microbial growth media such as casein soya bean digest agar and a single temperature within a 20 to 35 degree C range, plus or minus 2.5 degrees, should suffice.
Pre-Use, Post Sterilization Integrity Test of Sterilizing Filters (PUPSIT)
PDA recommends case-by-case comprehensive risk assessments on whether to perform pre-use, post-sterilization integrity testing of sterilizing grade filters. The assessment should be done by line and by product, PDA says.
Such testing can prevent risk of contamination, but it also can introduce contamination. The bottom line for PDA is to avoid such testing if it increases overall risk to product quality.
The effect on business risk should only be considered in cases where testing doesn't increase the quality risk.
Use of Two Sterilizing Grade Filters for Product Sterile Filtration
PDA says the authorities should not require redundant sterilizing filters in series, but that use of filters in parallel can be appropriate. Regardless, sterilizing filters should be placed to minimize the number of downstream aseptic connections.
Methods of Production Requirements for Water for Injection
PDA recommends allowing the use of any validated method such as reverse osmosis for achieving sufficient quality for water for injection.
The European Pharmacopoeia only accepts the distillation method.
Gori cautioned the audience against adopting the PDA recommendations prematurely. “You still need to comply with the European requirements for the European markets, unless you have some investigator who's using common sense and accepts our interpretation.”