首页
>
资讯
>
2022年5-6月FDA和EMA药品申请和补充申请报告
出自识林
2022年5-6月FDA和EMA药品申请和补充申请报告
2022-07-15
根据识林药品数据库,在2022年5-6月期间美国FDA和EMA集中审评的药品审评审批情况,包括补充申请,情况如下:
FDA批准了24个NDA (其中1个为计划批准)、3个BLA (CDER批准2个,CBER批准1个)和155个ANDA (其中19个为计划批准):
批准第二款用于治疗遗传性转甲状腺素蛋白淀粉样变性多发性神经病(hATTR-PN)的双链小干扰核糖核酸Amvuttra,靶向突变型和野生型转甲状腺素(TTR)信使RNA(mRNA),其正义链的3’端链接三个N-乙酰半乳糖胺(GalNAc)残基的配体,以使siRNA能够传递到肝细胞。
批准Mounjaro作为饮食和运动的辅助手段,用于2型糖尿病成人患者控制血糖。Mounjaro选择性地结合并激活 GIP 和 GLP-1 受体。Mounjaro以葡萄糖依赖性方式增强一相和二相胰岛素分泌,并降低胰高血糖素水平。
FDA共批准了591个补充申请 :
CAR-T 产品Breyanzi提线,用于特定B细胞淋巴瘤的2治疗。
撤销Rubraca适应症:单药用于治疗接受两种或两种以上化疗的有害BRCA突变(生殖系和/或体细胞)相关的晚期卵巢癌患者。
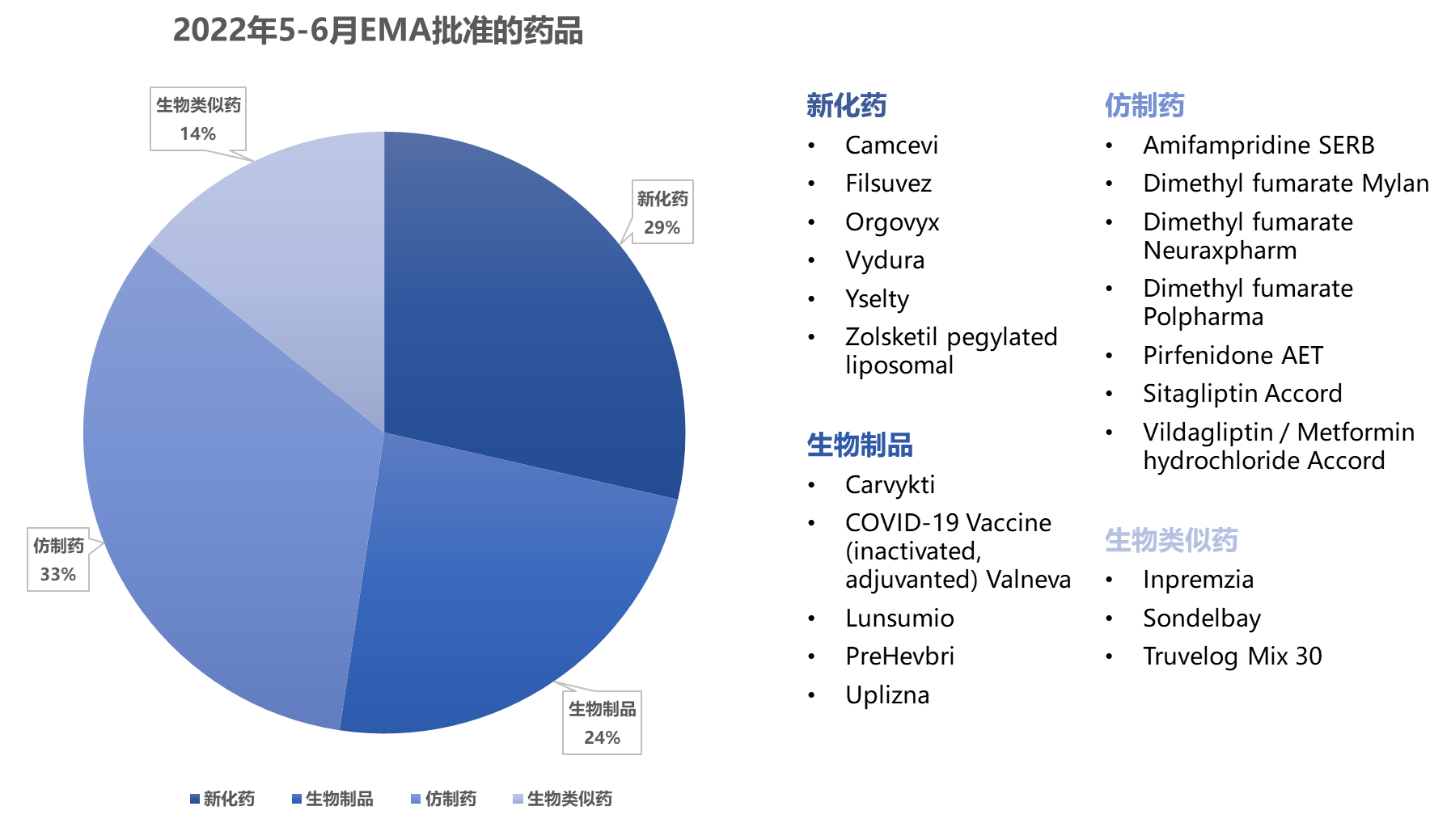
EMA通过集中审评批准了6个新化药、5个生物制品 、7个仿制药 和3个生物类似药 ,拒绝批准一个产品。
批准植物药(桦树皮的提取物)Filsuvez用于6个月及以上患者伴营养不良的交界性大疱性表皮松解症的局部皮层伤口。
批准欧盟首款针对新冠病毒的灭活疫苗,是继阿斯利康、辉瑞、Moderna、强生和Novavax之后的第六种新冠病毒疫苗。
拒绝批准Ipique(贝伐珠单抗)拟用于治疗新生血管(湿性)年龄相关性黄斑变性(AMD),该产品计划说明书中的安全性信息部分与Avastin相同。
EMA更新了266个药的850个变更申请信息。
批准Kymriah(CAR-T)用于治疗进行过两线或多线系统治疗后复发或难治性滤泡性淋巴瘤的成年患者。
以下为简要内容,详细内容请登陆识林>研发注册>案例解析阅览报告全文 。
1. FDA批准的新药申请
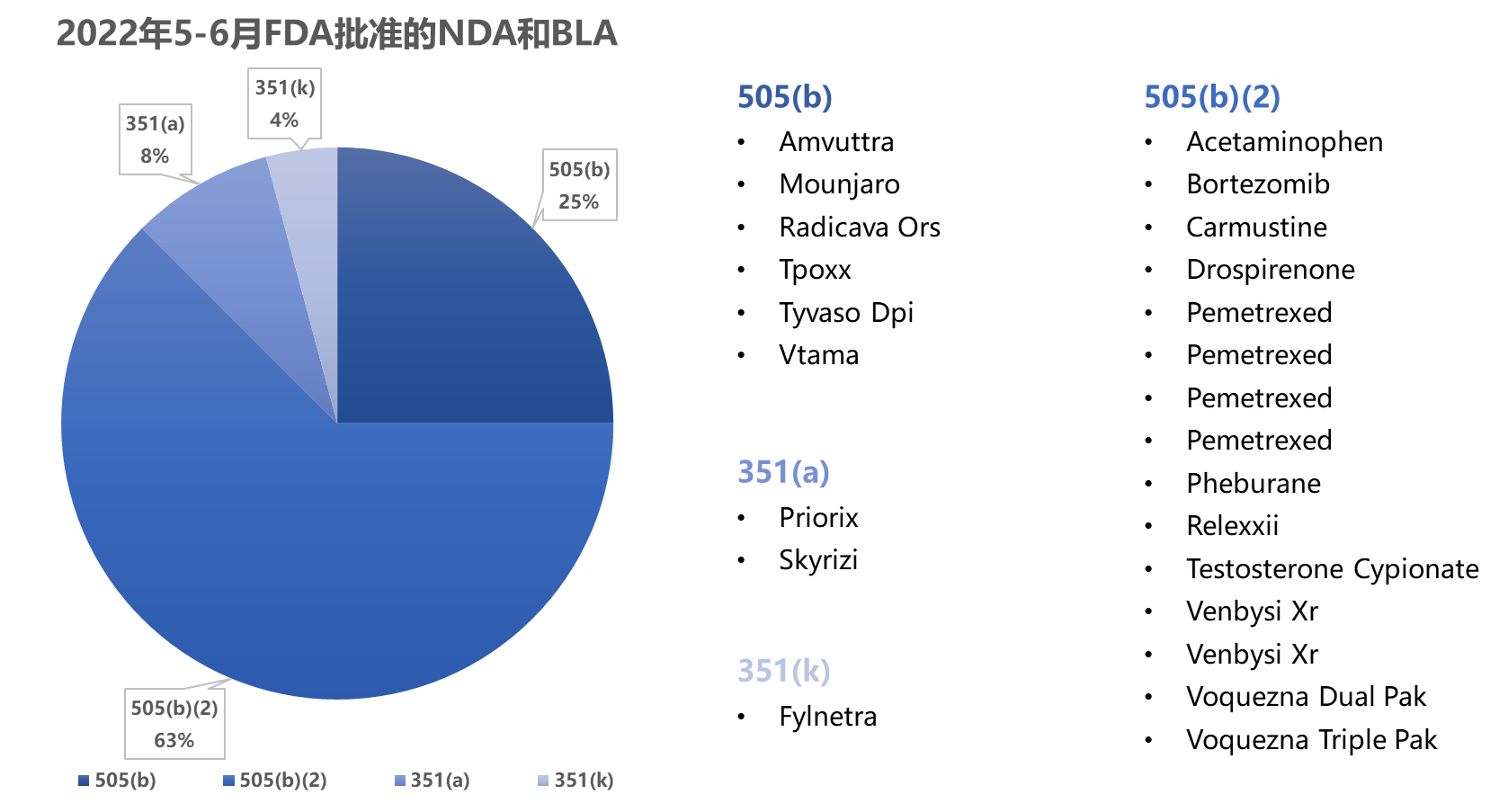
5-6月,FDA批准了24个NDA (其中1个为计划批准1 )、3个BLA (CDER批准1个351(a) 和1个351(k),CBER批准1个351(a))和155个ANDA2 (其中19个为计划批准)
505(b) 3 :6个,505(b)(2) 4 :15个,351(a) 5 :2个,351(k) 6 :1个
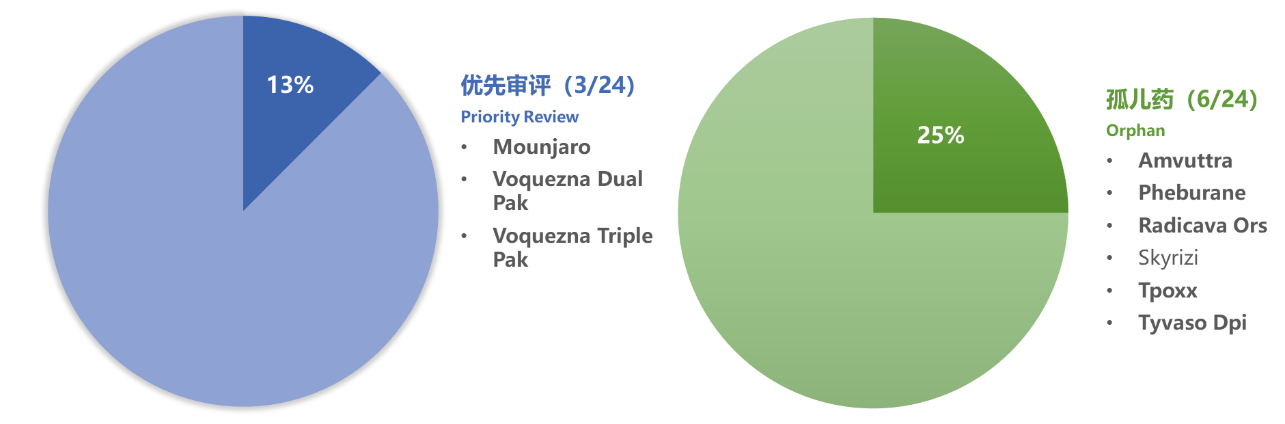
1.1 FDA优先审评和孤儿药7,8,9
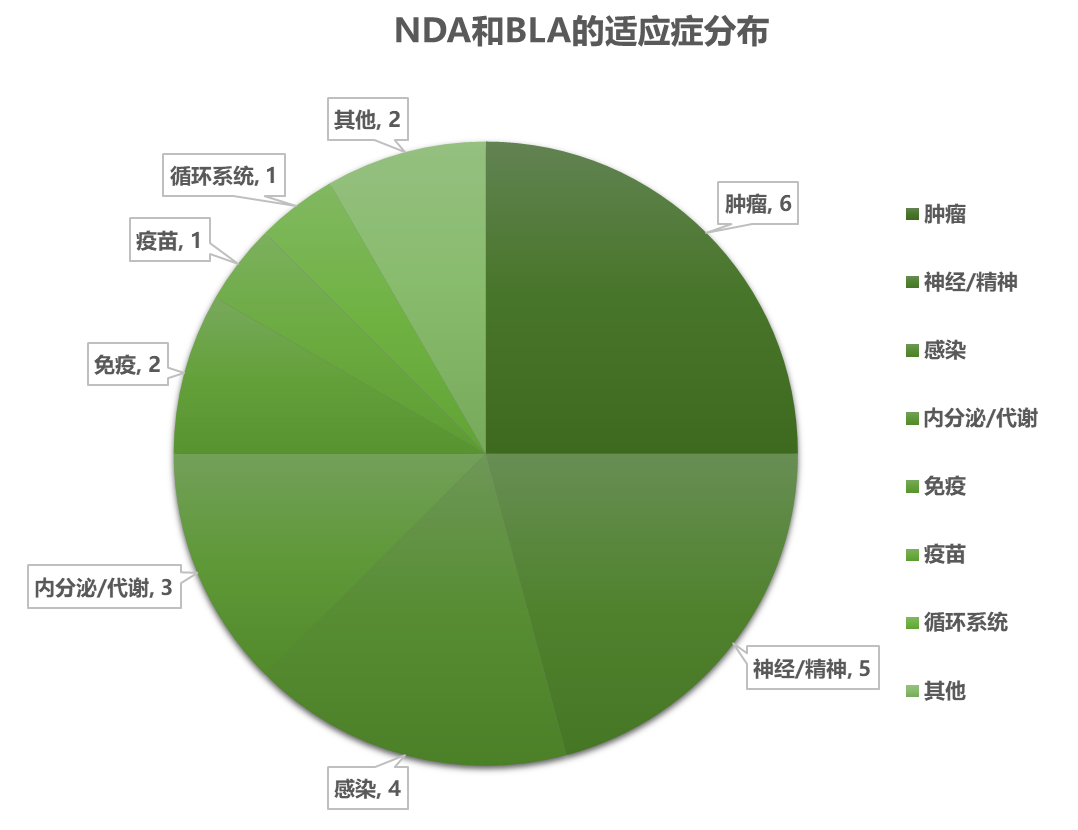
1.2 新批准药品的情况
2. FDA批准的补充申请
2.1 ANDA补充申请
共批准了178个ANDA 产品的333个补充申请 10 ,其中包含284个标签变更 、31个REMS 变更和18个CMC 变更。
2.2 NDA和BLA补充申请
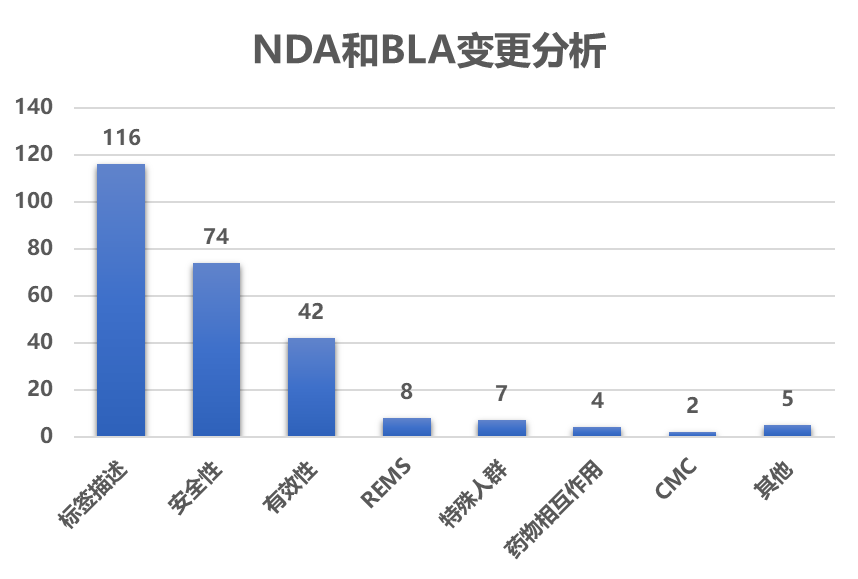
共批准了137个NDA 或BLA 产品的258个补充申请11 。
* 安全性:更新标签中黑框警告、警告和注意事项、禁忌症或不良反应 部分信息。
* 标签 描述:修改描述的文字,保证标签中前后内容的一致,505(b)(2) 产品与参比制剂的信息一致,修改标签微小错误;按照21 CFR 201.56(d) 和 201.57 ,修改表描述以合规。
* 有效性:新增适应症 ,扩增适应症人群,基于临床结果修改适应症描述,基于上市后研究或其他临床试验修改对适应症有效性部分描述等。
* CMC :包括修改剂型 、包装规格 、有效期 变更等。
* 特殊人群:有关儿童、老年人、妊娠、哺乳、肝肾功能不全患者等相关信息更新。
* 药物相互作用 :与其他药物、食物等显著相互作用的信息更新。
* REMS :REMS控制策略的变更 。
* 其他:包括标签中剂量调整、临床药理、非临床毒理等变更。
3. EMA批准和拒绝的新药
EMA集中审评批准了6个新化药、5个生物制品 、7个仿制药 、3个生物类似药 ,拒绝了1个产品的申请。
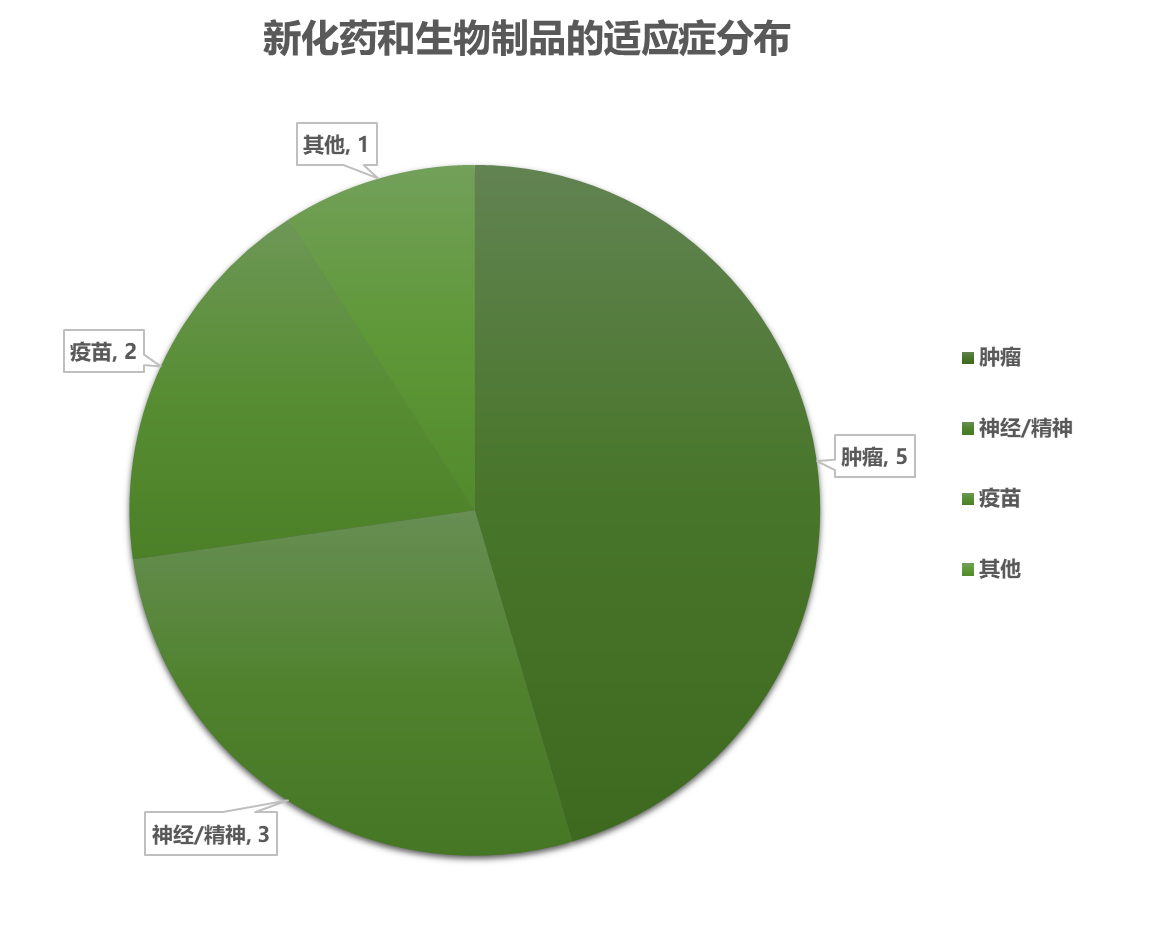
3.1 新化药和生物制品的情况
3.2 新化药 / 生物制品
4. EMA批准的变更
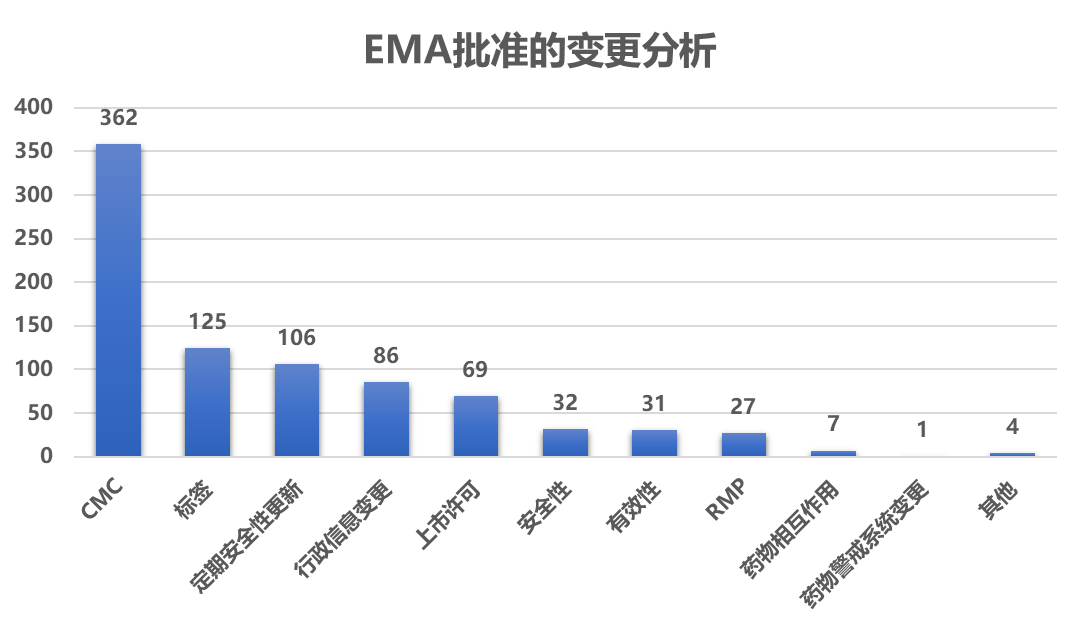
5-6月,EMA更新了266个药的850个变更 申请信息12 。
* 安全性:更新标签中警告和注意事项、禁忌症或不良反应 部分信息。
* 定期安全性更新:根据欧盟Regulation (EU) 1235/2010 、Directive 520/2012 和Regulation(EU) 520/2012 ,MAH 必须提交定期安全性更新报告(PSURs) 。
* 标签描述:修改描述的文字,保证标签内容的一致,与参比制剂的信息一致,修改标签微小错误,根据研究等更新标签内容等。
* 有效性:新增适应症,扩增适应症人群,基于临床结果修改适应症 描述,基于上市后研究或其他临床试验修改对适应症有效性部分描述等。
* CMC :包括修改剂型 、包装 规格、有效期 、工艺 、检测、批量等。
* 药物相互作用 :与其他药物、食物等显著相互作用的信息更新。
* RMP:风险管理 计划(RMP)的变更。
* 上市许可 :上市条件变更、上市许可更新和上市许可转移。
* 行政信息:MAH、生产商(生产、包装、检测、进口等)和供应商等的名字、地址、电话和邮箱等行政信息变更。另外,生产场地删除也记入行政信息变更。
* 撤市:监管机构要求或MAH 主动撤回产品。
* 其他:包括标签中剂量调整、临床药理、非临床毒理等信息更新,以及未明确说明的变更。
4.1 有效性
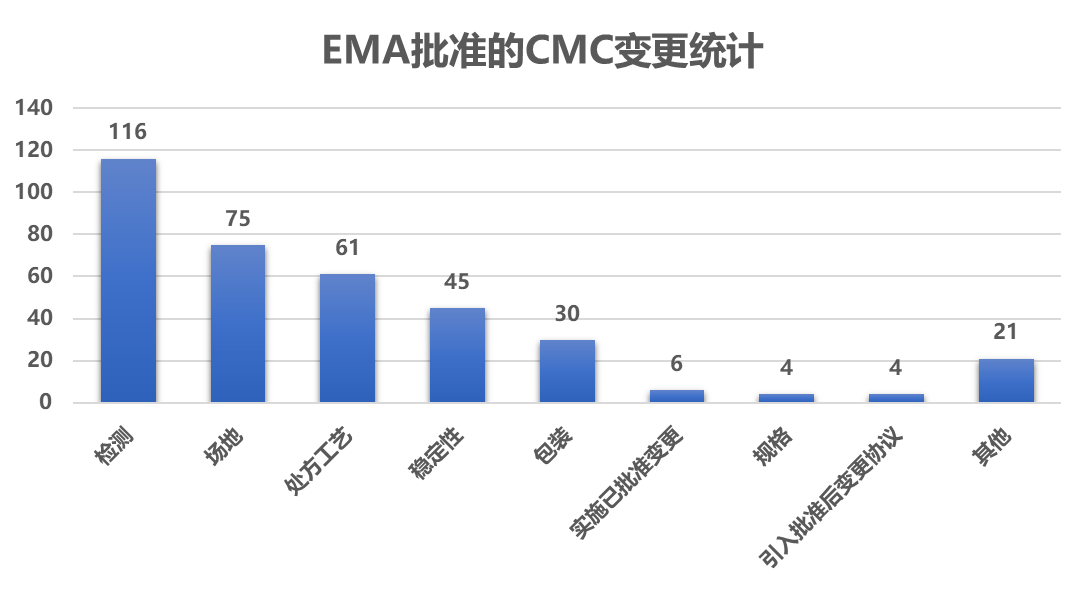
4.2 CMC
5-6月中有266个产品更新了变更信息,其中57.5%(153/266)的产品发生了 CMC 相关的变更 ,包括:
场地/生产厂商,包括生产、包装、进口、检测和批放行的场地/生产商
实施已批准的变更 :实施已批准的变更管理协议中预见的变更
4.3 撤市
5-6月期间,有5个产品因为商业原因,企业撤销了已批准上市的产品。
1 计划批准(Tentative Approval):计划批准的NDA 或ANDA 符合FD&C法案的批准要求,但是由于参比制剂的专利或转营期问题不能在美国销售。具体参考MAPP 5220.2 和【识林资讯-质量对话】ANDA 批准降为计划批准(Tentative Approval)
2 505(j) :仿制药 ,包含的资料表明申请的药品在活性成分、剂型 、规格、给药途径 、标签 、质量、性能特征和预定用途等方面,都与已批准的药品一致。
3 505(b) :批准信中标注为按照505(b)批准的产品,未明确说明是(b)(1)还是(b)(2)。
4 505(b)(2) :包含全部安全性和有效性研究报告的申请,但其中至少一部分批准所需的资料不是来自申请方所组织的研究,或申请方对这些资料没有引用权。
5 351(a) :包含全部安全性和有效性研究报告的生物制品 申请。
6 351(k) :生物类似药 , 该生物产品与参比制剂高度相似,允许在临床上无活性成分方面存在细微差别 , 并且该生物产品与参比制剂之间在安全性 , 纯度和效价方面均无临床意义上的差异。
7 优先审评 :Priority review,上市申请审评时间更短(6个月,而标准审评为10个月)。①治疗严重病症的药品申请 (原始申请或疗效补充申请),如果批准,能在安全性与有效性方面带来改善,或②按照505A 要求的儿科研究报告提出的标签 说明变 更补充申请,或③已被认定作为合格的感染性疾病治疗药品的申请或④任何带有优先审评凭单的药品申请或补充申请。详见FDA指南“Expedited Programs for Serious Conditions––Drugs and Biologics ” 和【识林主题词】-加快审评
8 孤儿药 :Orphan drug,美国2002年通过的《罕见病法案》将罕见病定义为在美国患病人数低于20万人,或患病率低于1/1500的疾病或病变。详见【识林主题词】-孤儿药 。
9 此处统计的是NDA 和BLA 中优先审评或孤儿药比率。
10 一个申请号视为一个ANDA 产品,一个补充申请 编号视为一个变更 申请
11 一个申请号视为一个NDA 或BLA 产品,一个补充申请 编号视为一个变更 申请,一个变更申请中可能包含多个变更类型标签 。不包括CBER批准的补充申请。
12 5-6月更新的变更 信息中会包含更早批准的变更内容;EMA 会同时更新一个药多个变更申请信息;一个变更申请中可能包含多个(种)变更信息。
识林® 版权所有,未经许可不得转载
岗位必读建议 研发(R&D) :关注加速审批路径和突破性疗法的资格标准,以指导新药开发。注册(Regulatory Affairs) :熟悉快速通道、突破性疗法、加速审批和优先审评的申请流程和时间节点。临床(Clinical) :了解临床试验设计和执行中的监管要求,确保数据满足监管标准。市场(Marketing) :注意加速审批产品的宣传材料需提交FDA审批。质量保证(QA) :确保生产和产品质量符合FDA的监管要求。文件适用范围 本文适用于美国FDA监管下的药品和生物制品,包括创新药、生物类似药、原料药等。主要针对治疗严重或危及生命疾病的药物,包括化学药、生物制品、疫苗等。适用于Biotech、大型药企、跨国药企、CRO和CDMO等各类企业。
文件要点总结 1. 加速审批程序概述 强调了加速审批路径旨在快速提供治疗严重或危及生命疾病的新药。 2. 突破性疗法资格标准 明确了突破性疗法的资格标准,包括对现有疗法的显著改善和初步临床证据。 3. 优先审评资格标准 规定了优先审评的资格,要求药品在安全性或有效性上有显著改进。 4. 加速审批的条件和后市场要求 详细说明了加速审批的条件,包括对替代终点的合理预测和后市场确认性试验的要求。 5. 沟通与合作 鼓励申办方与FDA在药物开发过程中进行早期和频繁的沟通,以确保满足监管要求。 以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
“ANDA”岗位:必读。需了解ANDA批准转换为暂时批准的政策和程序,确保在法院命令下合规操作。 “专利和独占权”岗位:必读。需评估法院命令对ANDA专利认证的影响,决定是否转换批准状态。 “法律和监管支持”岗位:必读。负责审查“转换为ANDA暂时批准”信函,确保法律文件的准确性。 工作建议:
ANDA岗位:密切关注法院命令和专利诉讼进展,及时与专利和独占权岗位协调,确保ANDA状态更新。 专利和独占权岗位:评估ANDA的专利认证,与法律和监管支持岗位合作,确定是否需要转换ANDA批准状态。 法律和监管支持岗位:审查所有相关文件,确保ANDA转换为暂时批准的决策和文件符合法律要求。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
注册 :必读。需理解法规对新药申请及市场独占期的影响。研发 :必读。指导儿科药物研究的设计和执行。市场 :必读。影响市场策略,尤其是涉及儿科用药的产品。临床 :必读。涉及儿科临床研究的设计和执行。QA :必读。确保研究和市场独占期符合法规要求。工作建议:
注册 :关注市场独占期的延长条件,及时与FDA沟通儿科研究要求。研发 :设计儿科研究时,考虑种族和民族多样性的代表性。市场 :评估儿科研究结果对市场策略的影响,准备相应的市场推广材料。临床 :确保儿科研究遵循FDA指南,并及时报告不良事件。QA :监督儿科研究的执行,确保符合FDA的质量和安全要求。适用范围:
要点总结:
儿科研究定义 :明确了儿科研究包括针对儿童群体的至少一项临床研究。新药市场独占期 :规定了在满足特定条件下,新药的市场独占期可延长至五年半。已上市药物市场独占期 :已上市药物在完成儿科研究后,其市场独占期同样可延长。儿科研究的执行 :FDA可以要求申办方开展儿科研究,并规定了响应时间。不良事件报告 :规定了儿科用药不良事件报告的流程和责任。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
PV(药物警戒)专员/负责人 :必读。负责确保药物警戒系统主文件(PSMF)的准确性和及时更新,以及药物警戒活动的合规性。QA(质量保证) :必读。监督药物警戒质量体系的建立和执行,确保符合规定要求。注册部门 :必读。了解药物警戒要求,以支持药品注册和监管合规。研发部门 :必读。在药物开发过程中考虑药物警戒要求,特别是在风险管理计划和后期授权安全研究方面。文件适用范围:
文件要点总结:
药物警戒系统主文件(PSMF) :明确了PSMF的结构、内容、维护、文件形式、分包和可用性等要求,以确保药物警戒系统的透明度和可追溯性。质量体系要求 :规定了药物警戒活动的质量体系的最低要求,包括人力资源管理、合规管理、记录管理和审计。Eudravigilance数据库监测 :强调了对Eudravigilance数据库的持续监测要求,包括识别新风险和变化风险、信号管理流程和工作共享。术语、格式和标准使用 :规定了在药物警戒活动中使用国际公认的术语、格式和标准,以促进信息交换和系统互操作性。风险管理计划和定期安全更新报告 :要求制定风险管理计划,并定期更新安全信息,以监控药品的风险-效益平衡。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。