|
首页
>
资讯
>
国内药政每周导读:中检院拟修订注册检验程序,CDE更新电子申报资料软件,专利登记可提交法律文书,中药特征图谱和稳定性研究,等
出自识林
国内药政每周导读:中检院拟修订注册检验程序,CDE更新电子申报资料软件,专利登记可提交法律文书,中药特征图谱和稳定性研究,等
2024-03-04
上周国内药政导读
【创新药与临床开发】
3.1,【CDE】关于发布《生长激素制剂用于生长激素缺乏症临床试验技术指导原则》的通告(2024年第18号)
目前已有多种 GH 制剂获批用于生长激素缺乏所引起的儿童生长缓慢和成人 GHD 的治疗,但是大部分为短效 GH 制剂,需要每日一次注射治疗,给长期治疗的依从性带来挑战,因此长效 GH 逐渐成为国内外生长激素领域的研发热点。目前有多种技术可以延长生长激素作用,包括微球缓释剂型、聚乙二醇化、前体药物、具有非共价白蛋白结合特性的生长激素衍生物、生长激素融合蛋白等。
为科学引导和规范我国 GH 制剂的临床研发,在借鉴国内外相关技术指导原则的基础上,结合临床试验进展和国内外相关指南制定本指导原则。
3.1,【CDE】关于发布《小儿便秘中药新药临床研发技术指导原则(试行)》的通告(2024年第19号)
本技术指导原则适用于“中医药理论、人用经验和临床试验相结合的中药注册审评证据体系”(以下简称“三结合” 中药审评证据体系)下的小儿便秘中药新药研发,旨在指导申请人在将临床经验方或医疗机构中药制剂开发为中药新药的过程中,基于中医药理论和人用经验制定适宜的研发策略,明确用于支持疗效评价的关键信息。鼓励将真实世界研究、适应性设计、以患者为中心的药物研发理念等应用于小儿便秘中药临床疗效评价,探索符合中医药疗效特点的儿童用药临床评价新工具,如患者报告结局、观察者/监护者报告结局等临床报告结局指标。
【CMC与仿制药】
2.27,【药典会】2024年度国家药品标准提高课题拟立项目录公示
公示期仅7日,截至3月5日。
相比药典公示,药企从这个目录能更早知晓药典即将到来的变化,并早做准备。
目录分为“品种”和“通用技术要求”两部分。
其中,“品种”里包含化药55个,生物制品7个(包括替雷利珠单抗和特瑞普利单抗),辅料34个,中药36个。
“通用技术要求”包含理化分析9个,生物检定8个,微生物10个等等。
建议分析人员查阅了解。
2.27,【CDE】关于发布《中药制剂特征图谱研究技术指导原则(试行)》《中药制剂稳定性研究技术指导原则(试行)》的通告(2024年第16号)
两文曾于2023年10月征求意见。
中药制剂迎来更多更细的药学指导原则。
中药制剂特征图谱系指中药制剂样品经适当处理后,采用适宜的分析方法,研究建立的能够反映多组份信息并体现其质量特征的图谱。中药制剂特征图谱对于识别中药制剂关键质量属性,研究量质传递,评价中药制剂质量的均一性、稳定性,提高中药制剂整体质量控制水平具有重要意义。
中药制剂稳定性研究应贯穿于药品的全生命周期,不应当止于其研究与开发过程,上市后还应当持续关注可能影响其质量的因素。本技术指导原则适用于中药制剂申请临床试验、上市许可、上市后变更等研究过程中的稳定性研究;中药制剂中间产物、天然药物稳定性研究亦可参考本技术指导原则。
【注册与审评审批】
2.29,【CDE】关于更新电子申报资料制作软件的通知
CDE根据近期收集的业界反馈意见以及电子申报资料使用中存在的问题,经充分评估后,对2023年12月11日发布的电子申报资料制作软件进行了优化更新,以进一步提高电子申报资料制作软件的易用性,提升用户体验。
本次更新内容主要包括增加文件数量统计和同名文件提示功能,完善临床试验数据库子文件夹创建功能,优化扩展文件夹命名规则,以及结合实际业务需要,细化软件中电子申报资料类型和对应的文档结构。
用户可阅读识林向导@贰雯撰写的《【解读】2024年3月电子申报新规验证标准、法规要求及相关技巧-V1.0》一文,作为实操参考。
【GXP与检查】
2.27,【中检院】公开征求《药品注册检验工作程序和技术要求规范(修订草案征求意见稿)》意见的通知
本文曾于2020年5月征求意见,2020年7月即落地。这次的修订草案征求意见截至3月27日。
注册检验,和注册核查一样,是上市(以及重大变更)的必经步骤,任何监管要求都可能影响品种批准的进度。
建议用户使用“页面比对”制作花脸稿,仔细思考修订内容背后的监管思路。下面是花脸稿里的一些修订示例:
- 新增提醒“申请人可向具备条件的药品检验机构提供电子化的注册检验用资料和样品标签。”
- 补充申请也可申请前置,“对于补充申请之前提出前置注册检验的,申请人应当对照药品上市后变更技术指导原则进行评估,与药品审评中心充分沟通后提出申请。”
- 资料方面,“如为非无菌化学药品或原料药,应提供微生物限度控制策略(包括申报质量标准中是否订入微生物限度项目;微生物限度的检验策略是逐批检测还是定期检测;其他必要的风险评估资料)。”
【政策法规综合】
2.26,【CDE】关于在中国上市药品专利信息登记平台设置法律文书提交模块的通知
自本通知发布之日起,专利权人、利害关系人或仿制药申请人可直接在中国上市药品专利信息登记平台提交“设置等待期申请书”、受理通知书、判决书、决定书或和解书等,无需再通过公文进行提交,之后可直接在“申请人之窗”中查看进度及处理结果。
此前已通过公文形式提交资料,且相关仿制药尚未完成审评的,需要在中国上市药品专利信息登记平台中再次进行提交。
【新药批准和报产】
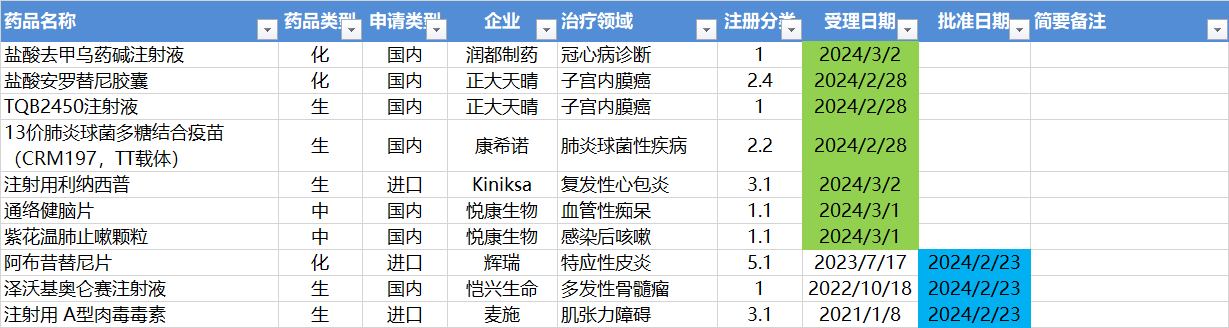
2.26-3.3,NMPA发布3个新药批准,CDE受理7个NDA
注:仅列出新药(包括改良型新药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
识林®版权所有,未经许可不得转载
必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |