|
首页
>
资讯
>
【研究】FDA 在新药审评中使用的患者经验数据的分析
出自识林
【研究】FDA 在新药审评中使用的患者经验数据的分析
2019-11-27
“患者经验数据”(Patient experience data, PED)是指与患者的经验、观点、需求和优先重点相关的有意义数据的系统性收集。PED 可以在 FDA 对产品申请的审评中扩充传统临床试验数据。
10 月 9 日发表在 DIA《治疗创新与监管科学》杂志上的一篇研究文章分析了 FDA 在 2018 年批准的 59 个新分子实体(NME)的审评文件中报告的 PED 的使用,发现在 59 个 NME 中,有 48 个的审评文件中包括一张总结在 FDA 药品审评期间是否使用了 PED 的表格,其中 34 个药在药品审评中使用了 PED。患者报告的结果(patient-reported outcomes, PRO)是 PED 最重要的来源,在 60.4% 的获批药品审评中使用。此外,研究还介绍了按 FDA 审评部门和按 FDA 监管认定归类的 PED 的使用。
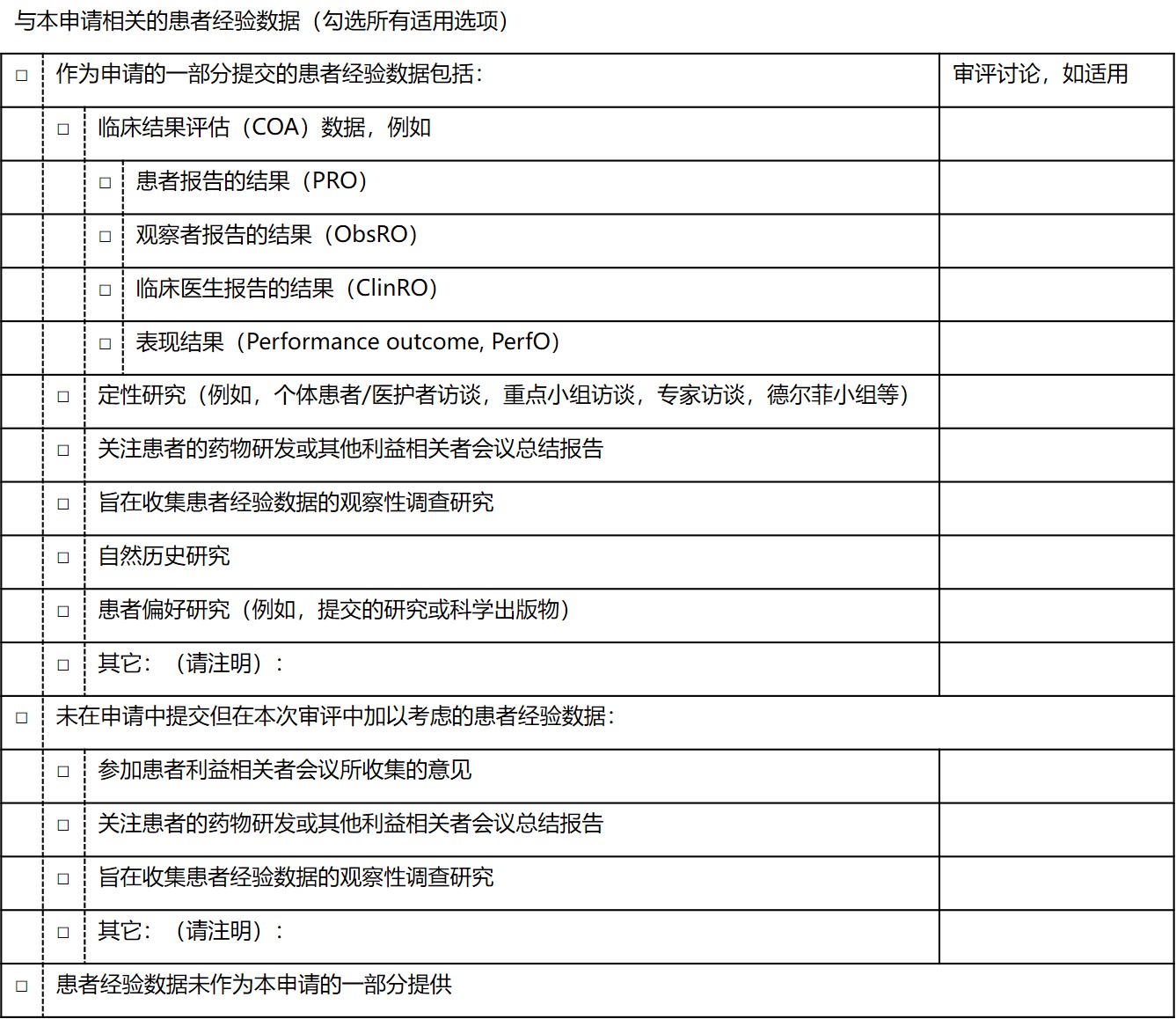
2016 年《21 世纪医药法案》第 3001 条要求 FDA 对于在药品申请批准中考虑的 PED 做出公开声明。FDA 通过在审评文件中包含一张索引所考虑的 PED 类型的表格(PED 表)来满足第 3001 条的要求(见下表)。PED 表包括 18 个单独的复选框,引用了申办人在提交的申请中包括的 9 种形式的 PED,以及未由申办人提交但 FDA 在审评中会加以考虑的 3 种 PED 形式。
在 FDA 2018 年批准的 59 件药品申请中,有 48 件(81.4%)在审评文件中包含 PED 表。11 件审评文件内不包含 PED 表的申请中,有 7 件申请是在 2017 年 6 月 16 日第 3001 条实施之前提交到 FDA 的,不需要公开报告 PED。48 个包含 PED 表的药品审评中有 34 个(70.8%)在审评中使用了 PED,其中 29 个(60.4%)药品的审评仅参考了申办人提交的数据,1 个(2.1%)药的审评仅参考了非申办人提交的数据,4 个(8.3%)药的审评既考虑了申办人提交的数据也考虑了非申办人提交的数据。申办人提交的 PED 主要以 PRO 的形式(29/48,60.4%)。
研究表示,与其它质量更高的 PED 来源相比,FDA 批准的药品审评更频繁地考虑 COA(尤其是 PRO)。研究分析的数据中,COA 占 PED 来源的近四分之三,而 PRO 是 PED 的最大单一来源。研究指出,鉴于 PRO 数据的定量性和在监管环境中更悠久的使用,与其它 PED 来源相比,FDA 和申办人对PRO 的使用都感到更加熟悉和易于使用。研究建议 FDA 和申办人应确保将除 COA 以外的所有的 PED 形式都适当地纳入到药品研发中,供 FDA 在审批时考虑。同样,研究还建议 FDA 考虑采取其它措施来增加 PED 来源的多样性。
这一研究分析是为了更好地了解 FDA 如何在监管决策中考虑 PED,这项分析应有助于建立有关 FDA 使用 PED 的基准,并可能有助于指导决策,以确保在未来的药物开发中充分听取患者经验。
作者:识林-Acorn
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
参考资料
[1] Kieffer, C. M., Miller, A. R., Chacko, B., & Robertson, A. S. (2019). FDA Reported Use of Patient Experience Data in 2018 Drug Approvals. Therapeutic Innovation & Regulatory Science.
岗位必读建议: - 研发(R&D):了解加速药品开发流程的新规定,确保研发项目符合新法规要求。
- 注册(Regulatory Affairs):掌握法规对药品注册流程的影响,优化注册策略。
- 临床(Clinical):关注临床试验设计的新指导原则,确保试验合规性。
文件适用范围:
本文适用于美国境内的化学药、生物制品、疫苗等药品类型,包括创新药、仿制药、生物类似药及原料药。主要面向Biotech、大型药企、跨国药企等企业类别。 文件要点总结: - 加速药品开发流程:强调了加速药品从发现到上市的整个流程,以促进21世纪医疗创新。
- 临床试验现代化:提出了对临床试验设计的现代化要求,以提高试验效率和患者参与度。
- 个性化医疗推进:鼓励发展个性化医疗方法,包括精准医疗和基因疗法。
- 数据共享与隐私保护:规定了数据共享机制,同时强调了患者数据的隐私保护。
- 监管框架更新:明确了对FDA监管框架的更新,以适应新兴医疗技术和产品。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |