|
首页
>
资讯
>
【周末杂谈】FDA 指南的新貌
出自识林
2023-04-16
为落实《简明文字法》,FDA开始为其监管指南配以图文并茂的快照
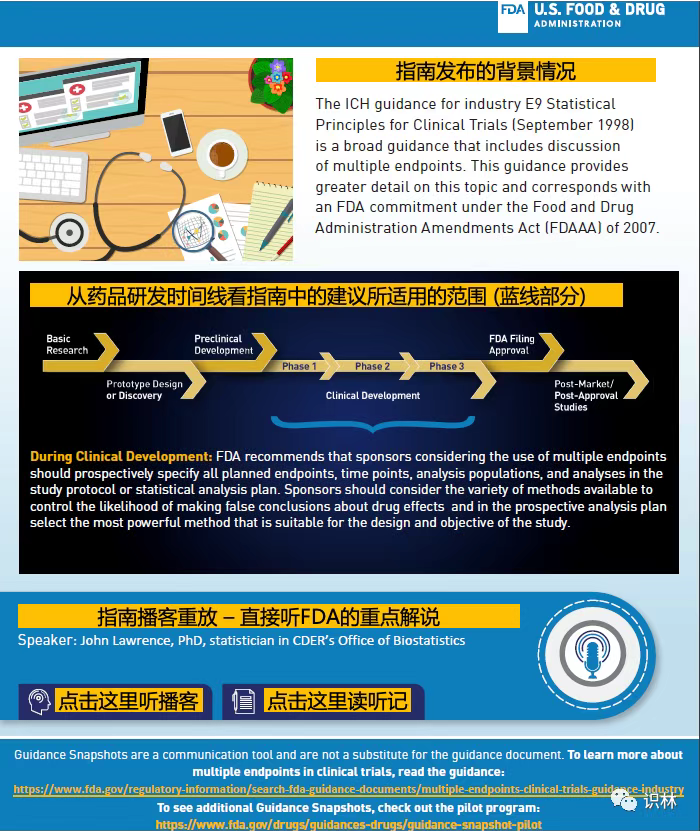
这周一,FDA公布了“使用创新的沟通方法来提高对“药品审评和研究中心(CDER)”指南的认知和理解:CDER 指南快照试点计划”。读过FDA指南的人恐怕都有过对整体用意和具体文字不甚明白的经历。FDA将为指南添加图文并茂的快照(snapshot),以便读者一目了然指南的内容、用途和来龙去脉,并配以播客重放和听记。
试点将先以下述两份指南起步:(1)“增强临床试验人群的多样性 - 资格标准、招募实践和试验设计”,及(2)“以患者为中心的药品开发指南 4:将临床结果评估纳入监管决策制定的终点”。这两份指南快照具体长成啥样子,尚不得而知,但可从FDA去年10月发布的一份指南快照中得到一些启发(其实,零星的快照早于 2018 年就有了,只是正式的试点计划刚开始)。如下面三张截图所示。
FDA的这项举措是为了落实于2010年生效的《简明文字法》(12年前?!),其目的是让所有联邦政府(不限于FDA)发布的公文易于公众理解和使用。请注意该法律中的如下三点:
- (1) 各政府部局要在其官网上设“简明文字专栏”,说明其所作所为。并给出专人的姓名和联络方式,以便公众问询和提意见。部局一把手还要在专栏上公布在简明文字方面工作的年报。
- (2) 规章(regulation)不受该法律管。规章本质上是行政法,其是否合法由法院解释。数百年的法律先例定义了行政法中字词和术语的含义,用通俗易懂的语言写规章会破坏法律先例的体系。
- (3) 该法律不能作为法律质疑的理由,因为质疑只会使政府公文回到传统的官僚术语,以模糊其含义来声称质疑是对公文的错误理解。换句话说,这有助于确保以易懂的语言解释公文的善意努力不会被用作质疑解释不清的基础。
这三点揭示了《简明文字法》的特殊用心,即,用透明和公众监督的方式来督促而不是强制官僚机构做善事。这是因为政府部门都愿意有所作为,但又都怕由此而引发风险。
笔者感谢北京大学访问学者Garth Boehm博士就上述三点给出的深入见解。
作者:榆木疙瘩
识林®版权所有,未经许可不得转载
适用岗位: - 临床(Clin):必读。在设计和执行临床试验时,需考虑如何扩大资格标准,以包含更广泛的参与者,确保试验结果更具代表性。
- 注册(Reg):必读。需了解FDA对于临床试验人群多样性的最新要求,以便在药品注册过程中更好地满足监管要求。
- 研发(R&D):必读。在药物开发的早期阶段,就应考虑如何通过试验设计和方法论吸引更多样化的参与者。
工作建议: - 临床(Clin):在设计临床试验方案时,应特别关注如何放宽入组标准,减少不必要的排除标准,以增加试验人群的多样性。
- 注册(Reg):在准备新药申请或生物制品许可申请时,应确保提交的数据反映了包括不同人群在内的广泛参与者,以满足FDA对多样性的要求。
- 研发(R&D):在药物开发的早期阶段,应考虑药物代谢和清除在不同人群中的差异,以及如何通过适应性设计和早期儿科发展计划来吸引更广泛的参与者。
适用范围:
本文适用于化学药和生物制品,包括用于治疗严重和危及生命条件或疾病的药物,由美国食品药品监督管理局(FDA)发布,适用于Biotech、大型药企、跨国药企以及CRO和CDMO等各类企业。 要点总结:
本文强调了在临床试验中增加人群多样性的重要性,提出了扩大资格标准、改善入组实践和试验设计的建议。文件中明确指出,应避免不必要的排除标准,确保试验人群更好地反映可能使用药物的人群。特别提到了在后期药物开发阶段扩大资格标准的重要性,以及在设计临床试验时考虑包括不同年龄、性别、种族和民族的参与者。此外,还讨论了如何通过减少参与者的负担、采用更具包容性的入组和保留实践,以及扩大访问权限来改善入组。对于旨在治疗罕见疾病或条件的临床试验,提出了特别的考虑,包括与患者倡导团体的早期接触和在早期试验中重新招募参与者。整体而言,文件鼓励赞助商考虑本文提出的方法,并根据需要开发其他方法。 以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位及工作建议: - 临床(Clinical):必读。在设计临床试验时,应考虑如何合理选择和报告多个终点,确保试验结果的科学性和合规性。
- 研发(R&D):必读。在药物开发过程中,需依据指南评估多个终点的合理性,优化临床试验设计。
- 注册(Regulatory Affairs):必读。在提交临床试验申请时,需确保终点选择和报告符合FDA指南要求。
适用范围:
本文适用于化学药和生物制品的临床试验,包括创新药、仿制药和生物类似药,由美国FDA发布,适用于跨国药企和Biotech公司。 文件要点总结: - 终点选择与报告:明确指出在临床试验中选择和报告多个终点时应遵循的原则和标准。
- 统计考量:强调了在多个终点分析中统计学考量的重要性,包括对多重比较的控制。
- 监管沟通:鼓励在临床试验设计阶段与FDA进行沟通,以确保终点选择和报告的合规性。
- 数据完整性:规定了在报告多个终点时,如何保证数据的完整性和透明度。
- 适应性设计:讨论了适应性设计在多个终点分析中的应用,以及其对试验结果的影响。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - 临床(Clinical):负责临床试验的设计、执行和数据分析,需理解COA在终点评估中的应用和意义。
- 研发(R&D):涉及药物开发过程中的患者体验数据收集和应用,需了解COA的选择和验证。
- 注册(Regulatory):负责药品注册文件的准备和提交,需掌握COA数据的格式和提交要求。
工作建议: - 临床(Clinical):
- 确保临床试验设计符合FDA指南,特别是在选择和验证COA时。
- 在试验中实施适当的COA,以确保数据的准确性和代表性。
- 研发(R&D):
- 与患者和护理人员合作,收集关于疾病影响和治疗偏好的数据。
- 根据患者反馈选择或开发适合的COA工具。
- 注册(Regulatory):
- 遵循eCTD和其他相关指南,准备和提交包含COA数据的注册文件。
- 与FDA沟通,确保提交的数据满足监管要求。
适用范围:
本文适用于化学药、生物制品和医疗器械等药品类型,涉及创新药、仿制药和生物类似药等注册分类,由美国FDA发布,适用于Biotech、大型药企、跨国药企以及CRO和CDMO等企业类别。 要点总结: - COA终点考量:强调了在临床试验中选择和构建基于COA的终点的重要性,包括对终点的合理性论证和统计分析的考虑。
- 治疗效益的意义评估:描述了如何评估基于COA的治疗效果对患者的意义,包括影响COA分数解释的因素和支持解释性的证据收集方法。
- 缺失数据处理:讨论了在COA数据中处理缺失数据的方法,强调了对缺失数据的敏感性分析。
- 研究设计其他考虑:包括盲法、实践效应、辅助设备使用等对COA数据收集和解释的影响。
- 数据提交格式:提供了关于如何格式化和提交COA数据以支持监管决策的指导。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |