|
首页
>
资讯
>
IPEM 课程 欧盟无菌附录对生物产品和疫苗的影响,分析方法在生物制药和质量控制上的应用
出自识林
IPEM 课程 欧盟无菌附录对生物产品和疫苗的影响,分析方法在生物制药和质量控制上的应用
2023-06-05
以下文章来源于IPEM ,作者IPEM
教师简介
Ian Thrussell先生,曾任世界卫生组织GMP、无菌药品检查专家,首席检查员。2006-2011年,作为英国药品与健康产品管理局 (MHRA) GMP检查员赴欧洲药品管理局 (EMA) 检查工作组工作,出任英国药品安全委员会 (UK CSM) 化学与药剂学分委会督查代表,被欧洲委员会(EC) 委任为ICH Q10专家工作组成员。曾赴波多黎各、日本、印度、美国、中国、波兰、罗马尼亚、澳大利亚、意大利开展GMP检查。曾获牛津大学埃克塞特学院化学硕士学位。
课程介绍
欧盟无菌附录已经不是新法规了,从2017的第一稿,到现在已经六年了。里面的大部分要求,也都是行业经验和已有监管要求的固化和细化。
Ian有三十年的检查经验,最近三年多半在中国,大部分是在生物产品和疫苗企业。他看到的大部分缺陷,是偏差调查不彻底,无菌保障不系统,质量体系不可靠等基本缺陷。
法规已经明确,缺陷也很基础。从字面差距分析看,欧盟无菌附录有超过半数的要求与我国现行无菌附录有差异或新增。但字面要求背后的考量,不同版本有哪些差别,收到了哪些反馈,为什么这么改,不那么改,这些就是Ian打算在课上分享的。同样关于缺陷案例,为什么总是出现同样的基本问题,与产品工艺的关联是什么,与产品工艺无关的是哪些,也是Ian希望和大家讨论的。
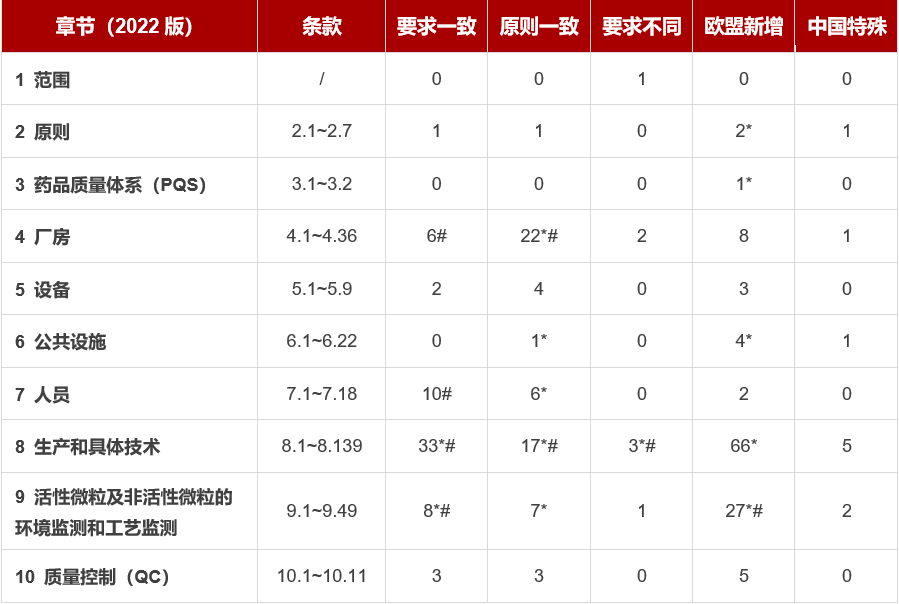
* 条款的差异分析中,部分条款内容相近,有合并;
# 部分条款有拆分,便于逐条对比;
课程大纲
第1节 - 新版GMP无菌附录1(EU&PICs)/附录2(WHO)的介绍
讨论新版无菌指南的一些关键要点,及其对不同产品(化药/生物药/疫苗)的无菌原料药和成品制剂的适用性。还将讨论实施新版无菌要求的挑战。
第2节 - 污染控制策略(CCS)
讨论实施有效CCS的关键要求,以及监管部门对已有产品和新产品CCS实操中的监管期望。
第3节 - 无菌产品检查中的代表性GMP缺陷领域
新版无菌附录的审慎草案发布迄今已有3年多,并且WHO检查员在检查中一直在运用这些新指导原则。我们发现了哪些问题,有哪些是旧问题?有哪些可能是新问题?您已经做好哪些准备了?
第4节 - GMP公开课专题讨论。案例研究和现场提问
邀请代表们匿名或当面提问进行讨论。问题可以在课程之前或在课间休息时提交,老师将与代表们讨论这些问题。
大家通常会有很多问题,但有时不愿意口头表述。鉴于此,可以在课程之前或在课程期间将问题提交给课程项目组,并就这些问题举例讨论。其目的是让代表们从彼此的经验和问题中学习,而不仅仅是让老师尽力提供答案!
第5节 - 有效的调查和CAPA
内毒素案例研究
蒸汽灭菌案例研究
教师简介
郭允籣女士,现任信达生物制药集团全球R&D运营负责人。曾先后在美国Amgen、Abbott和Genzyme等生物制药公司从事产品开发并担任产品负责人及CMC负责人,有25年以上从事蛋白分析科学与质量研究的丰富经验。曾经带领国内外多个单抗产品从临床前到BLA申报等一系列工作,熟知CMC开发及产品上市的一整套过程。从产业化的角度对新药研究、产品开发、蛋白质结构表征及特性研究、制剂研究、产品质量研究及质量标准的建立、质量控制、稳定性研究、技术转移、国内外IND及BLA申报等有着多方面的经验,包括不同国家监管机构对抗体和蛋白产品的审评审批过程:FDA、EMA、Health Canada、PMDA、TGA、MFDS、BPOM和NMPA等。曾担任美国CaSSS学会和MSB国际会议联合主席。曾荣获Amgen“杰出科学奖”、“最佳项目负责人奖”。曾是国家十二五项目负责人。毕业于美国麻省理工学院,曾在M.I.T.授课生物工程研究生课程,目前担任蛋白质质量联盟质量专家委员会主席。
课程介绍
从产业化角度,通过12个工业界实际案例解析,对不同的分析方法及其在生物制药领域中的应用进行阐述。以蛋白质药物的特性为基础,评估和确定生物制药中蛋白的关键质量属性CQA,建立评估CQA的分析方法,阐述分析方法与质量标准之间的关系。在产品生命周期的不同阶段,设计和开发不同的分析方法,对产品质量进行以阶段性为基础的有效控制;以案例分析的形式,对蛋白的纯度、杂质、一级结构、高级结构、结构异质性、生物学活性等重要属性进行剖析,解析蛋白结构与功能之间的关系,从而有效地制定产品质量的分析控制策略,以确保产品的质量控制、安全性和有效性。本课程还以工业界实例为基础,剖析分析方法验证中遇到的问题,采用DOE方法论,对分析方法的耐用性进行说明,从而达到分析方法的稳健性。
课程大纲
监管科学的理念
分析方法开发与产品特性研究
- 产品特性研究:纯度、杂质、异质性、生物学活性、Fc功能
CQA与蛋白结构表征
分析方法验证与质量控制
课程目标
了解各种分析方法的特点和用途,以及它们所检测的产品质量属性;了解分析方法开发中可能遇到的问题和需要掌握的重要要素,以及分析方法在整个生物药产品开发、工艺验证、上市阶段过程中所起的重要作用。对于产品的不同阶段,制定相应的产品质量控制策略,从而达到产品生命周期中的有效管理。
课外要求
阅读材料:《生物技术药物研究开发和质量控制》第3版 王军志院士
报名方式
点击链接或扫描下方二维码报名
电话:010-62750823
邮箱:register@ipem-prog.org
【IPEM2023年课程计划】点击查看
法规指南解读 适用岗位: 工作建议: - QA:确保所有生产活动符合GMP要求,监督无菌药品生产流程。
- 生产:按照GMP要求执行无菌生产操作,确保产品质量。
- 研发:在药品开发阶段考虑GMP合规性,设计符合要求的生产流程。
- 临床:确保临床试验用药的无菌性和质量符合GMP标准。
- 注册:在药品注册过程中提供符合GMP要求的生产和质量控制信息。
适用范围:
本文适用于化学药品、生物制品的无菌药品生产,包括原料药、制剂等。适用于欧盟地区的Biotech、大型药企、跨国药企等。 要点总结: - 无菌药品生产环境:强调了对无菌药品生产环境的严格控制,包括洁净室的分类和设计,以及对生产环境的持续监测。
- 质量风险管理(QRM):在整个文件中,QRM是确保无菌药品生产质量的核心原则,要求企业在设计和控制生产设施、设备、系统和程序时应用。
- 关键控制点:提出了无菌药品生产过程中的关键控制点,包括设施设计、设备操作、过程验证、环境监测和人员培训等。
- 污染控制策略(CCS):强调了CCS在无菌药品生产中的重要性,要求企业实施全面的CCS以确保产品质量和安全。
- 无菌工艺验证:要求对无菌工艺进行验证,包括无菌过程模拟(APS)和其他相关测试,以确保生产过程能够持续产生无菌产品。
以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读:ICH Q10 Pharmaceutical Quality System 适用岗位(必读): - QA:确保质量体系符合ICH Q10要求,监控质量体系的实施和持续改进。
- 注册:理解ICH Q10对药品注册的影响,确保注册文件与质量体系要求一致。
- 研发:在药品开发阶段应用ICH Q10原则,确保产品和流程的质量。
- 生产:根据ICH Q10要求,管理商业化生产过程中的质量控制和持续改进。
- 药物警戒:利用ICH Q10框架下的知识管理和质量风险管理,优化药物警戒活动。
工作建议: - QA应定期审查和更新质量手册,确保其反映ICH Q10的要求。
- 注册人员应确保所有注册文件和提交材料遵循ICH Q10的质量体系框架。
- 研发团队应将ICH Q10的原则整合到药品开发的每个阶段。
- 生产部门应依据ICH Q10建立和维护商业化生产的质量控制体系。
- 药物警戒部门应使用ICH Q10提供的工具进行风险评估和管理。
文件适用范围:
本文适用于支持化学药、生物制品及生物技术产品的开发和制造的系统,包括API和成品药,涵盖产品生命周期的各个阶段。适用于全球范围内的制药企业,包括Biotech、大型药企、跨国药企以及CRO和CDMO等。 要点总结: - 质量体系模型:“ICH Q10提供了一个基于ISO质量概念的综合性药品质量体系模型,与ICH Q8和Q9相辅相成。”
- 管理责任:“高级管理层有最终责任确保有效的药品质量体系的建立,以实现质量目标。”
- 持续改进:“ICH Q10鼓励使用科学和基于风险的方法,在产品生命周期的每个阶段促进持续改进。”
- 知识管理与质量风险管理:“知识管理和质量风险管理是实施ICH Q10并成功实现其目标的推动因素。”
- 监管方法:“ICH Q10的实施效果通常可以在生产场所的监管检查中评估。”
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - QA(质量保证):负责确保生物制品的生产、控制和测试符合GMP要求。
- 生产:负责按照GMP原则进行生物制品的生产活动。
- 研发:涉及生物制品的研发和工艺开发,需遵守GMP原则。
- 注册:负责将WHO指南转化为国家要求或进行合理修改。
- 质量控制(QC):负责生物制品的质量控制和测试,确保符合标准。
工作建议: - QA:定期审查和更新GMP程序,确保与WHO指南保持一致。
- 生产:在生产过程中实施质量风险管理(QRM),控制污染和交叉污染风险。
- 研发:在产品开发阶段考虑GMP要求,确保研发到生产的顺利过渡。
- 注册:与国家监管机构(NRA)沟通,确保WHO指南被正确采纳或修改。
- QC:根据WHO指南进行生物制品的质量控制和稳定性测试。
适用范围:
本文适用于生物制品的制造、控制和测试,包括化学药品、生物制品、疫苗等,适用于创新药和仿制药,发布机构为WHO,适用于Biotech、大型药企、跨国药企等。 要点总结: - GMP补充指南:强调了生物制品GMP的补充指南,包括种子库和细胞库的管理、生产和质量控制的特殊考虑。
- 质量风险管理(QRM):明确了QRM在生物制品生产中的重要性,特别是在控制策略的开发中。
- 人员健康与培训:规定了生产和控制人员的健康监测和培训要求,以确保产品安全和减少污染风险。
- 起始材料控制:强调了对起始材料的严格控制,包括供应商评估和材料测试,以保证产品质量。
- 环境控制与监测:详细说明了环境控制和监测的要求,特别是在生物制品生产过程中对特定微生物的检测。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |