关于数据可靠性,通常包括不完整、不一致或不准确的数据,Ashley 指出,过去四年向 API 生产商发出的警告信中,有 73% 涉及数据可靠性指证。他还引用了今年 8 月份发给 API 生产商 Lantech Pharmaceuticals 的警告信,信中指出,公司承认“常规永久删除三个月以上的回收溶剂气相色谱数据而不做任何备份。”Ashley 补充表示,“大家可以看到的数据可靠性问题只是冰山一角”,并指出,经常发生的错误会掩盖其它问题。【缬沙坦危机新进展:FDA 瞄准回收溶剂供应商 2019/08/20】
他还强调了 ICH Q7 指南,指南涉及 API 的CGMP,解释了“代理商、经纪人、分销商、再包装商或重新贴签商应将从 API 或中间体生产商那里接收到的所有质量或监管信息转给客户,并将从客户那里收到的信息转给 API 或中间体生产商。”
在杂质方面,他建议企业参考 ICH 指南 Q3A、Q3B(R2)、Q3C、Q3D 和 M7(R1),因为已经在一些不同的药品中发现存在亚硝胺杂质,并且已因此召回了降血压的沙坦类药物和治疗胃灼热的雷尼替丁。但是 FDA 最近对雷尼替丁药物中的杂质水平的态度有所淡化,认为杂质“与食用烧烤或熏制肉类等普通食品时所期望的含量相似。”
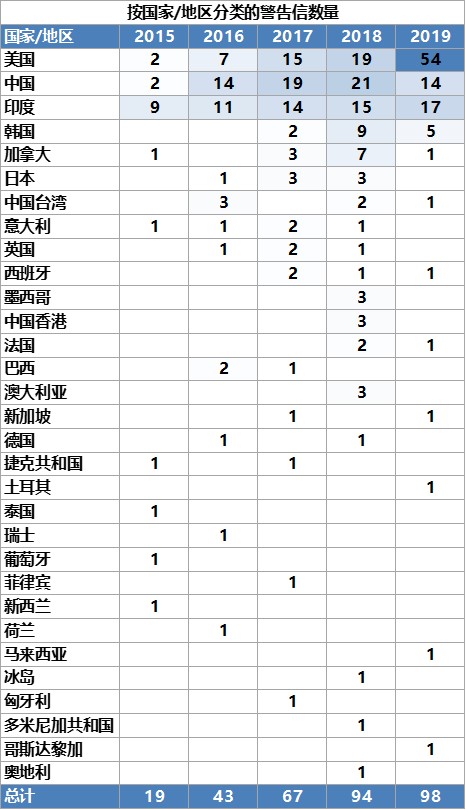
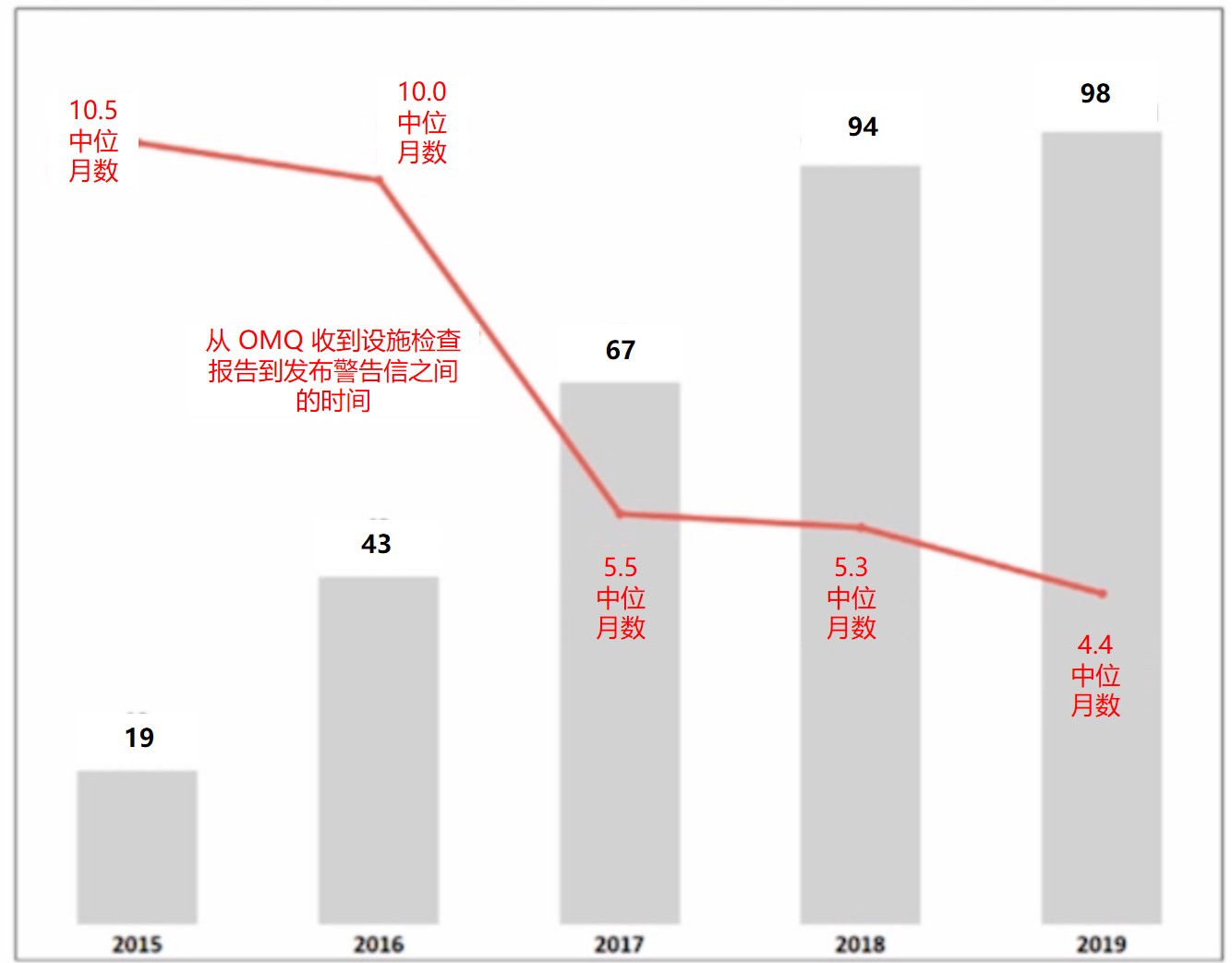
警告信数量增加,发布时间缩短
除对 API 的担忧外,Ashley 介绍指出,总体而言,从 2015 财年到 2019 财年,FDA 发出的警告信数量有所增加。2019 财年收到警告信最多的国家是美国,高达 54 封,其次是印度(17 封)和中国(14 封)。而前些年 FDA 对印度或中国发的警告信数量超出美国本土,目前尚不清楚这些变化是与国外检查数量有关还是因为美国公司的合规状态陷入低迷。根据合规办公室下的生产质量办公室主任 Francis Godwin 在最近的制药质量研讨会上的报告显示,发给原料药商的警告信数量与往年相比“相对持平”甚至有所减少,2017 财年为 21 封,2018 财年为 20 封,2019 财年为 13 封。
要点总结: ICH M7(R1)指南旨在为药物中的DNA反应性(基因毒性)杂质提供评估和控制框架,以限制潜在致癌风险。指南强调了在药物合成和降解过程中产生的基因毒性杂质的识别、分类、鉴定和控制。特别指出,对于已知的基因毒性致癌物,应基于致癌潜力和线性外推计算特定的可接受摄入量(AI),或使用国际监管机构已发布的值。对于表现出非线性剂量-反应关系或具有实际阈值的化合物,可基于无观察到效应水平(NOEL)和不确定性因子来计算每日允许摄入量(PDE)。指南还提供了基于治疗持续时间的不同可接受摄入量的调整方法,并讨论了多种控制策略,包括过程控制和定期检测。此外,指南还涉及了对上市产品的考虑,包括对药物合成、制剂变更和临床使用变更的评估。