《仿制药使用者付费修正案》(Generic Drug User Fee Amendments,简称GDUFA)是FDA与仿制药行业协商达成的一项协议,旨在通过收取用户费用来支持FDA对仿制药的监管活动。在GDUFA III中,FDA与行业达成了一项新的共识,即设立“警告信后会议”(Post-Warning Letter Meeting)机制,以帮助企业在收到警告信后更好地进行整改,并获得FDA的初步反馈。
根据指南,警告信后会议通常在企业对警告信作出初步回应后的6个月或更晚时间举行。但FDA也允许企业根据自身整改进度,申请提前召开会议。FDA会根据具体情况决定是否批准提前召开会议的请求。值得注意的是,即使企业参加了警告信后会议,FDA仍保留采取监管行动的权利,且会议中FDA提供的建议并不具有约束力(FDA advice provided at a Post-Warning Letter Meeting is not binding on the Agency)。
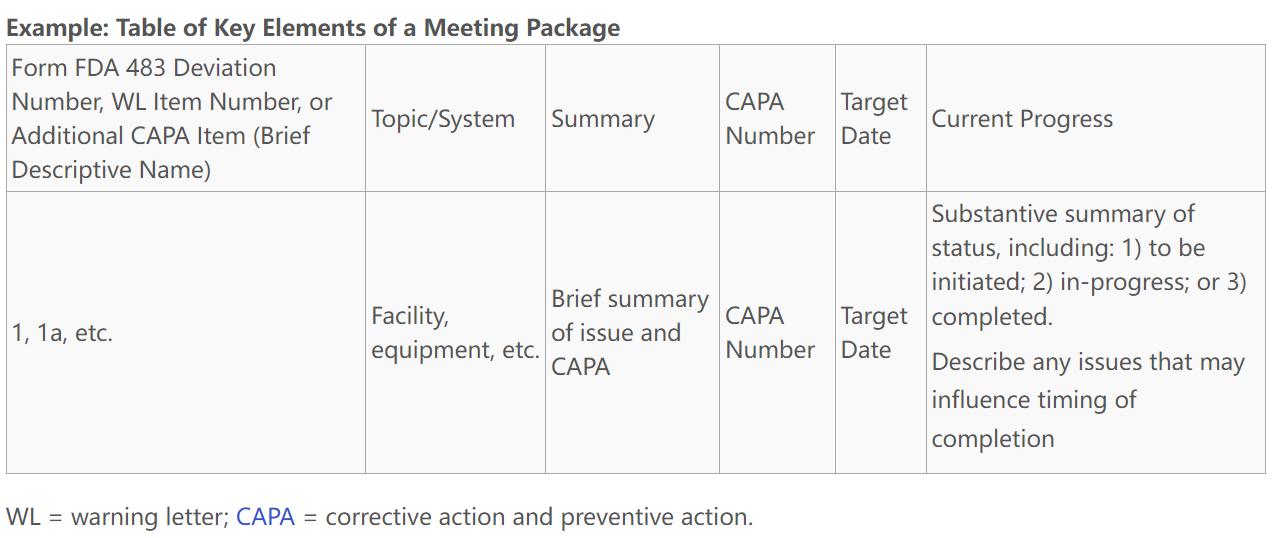
要点总结: FDA发布的《Post-Warning Letter Meetings Under GDUFA》指南,旨在规范仿制药企业在收到警告信后与FDA的会议流程。指南强调,只有在企业满足特定条件,包括CGMP合规状态为OAI、已支付GDUFA设施费用、且警告信内容仅限于与人类药品制造相关的违规时,企业才有资格请求会议。企业需提交完整的会议请求包,包括CAPA计划和补充信息,以证明已在系统性整改中取得合理进展。FDA将根据提交的CAPA计划的完整性和整改进展决定是否批准会议请求。若请求被批准,FDA将提供初步反馈;若被拒绝,企业将收到书面通知,并有机会在三个月后重新提交请求。此外,FDA可能推迟会议以进行重新检查。整个流程强调了企业在整改过程中与FDA沟通的重要性。