首页
>
资讯
>
FDA 仿制药论坛上报告的一些审批数据及申报建议
出自识林
2020-04-20
美国 FDA 于 4月15-16日举行了为期两天的仿制药论坛 ,会上有许多精彩话题,包括具体产品指南、仿制药办公室(OGD)标签审评和要求的介绍、简化新药申请(ANDA )与FDA的互动、电子提交、ANDA 计划绩效审查和改善 ANDA 质量的小贴士,以及根据行动函时间对一些申请的案例研究。另外还有关于仿制药药物警戒 、全球仿制药前景、ICH Q12 、常见 CMC (质量)问题以及如何避免、仿制组合产品 等非常有吸引力的话题。
今天我们先来看看 OGD 监管运营办公室主任 Ted Sherwood 所做的关于 ANDA 计划绩效审查演讲中的一些关键内容。
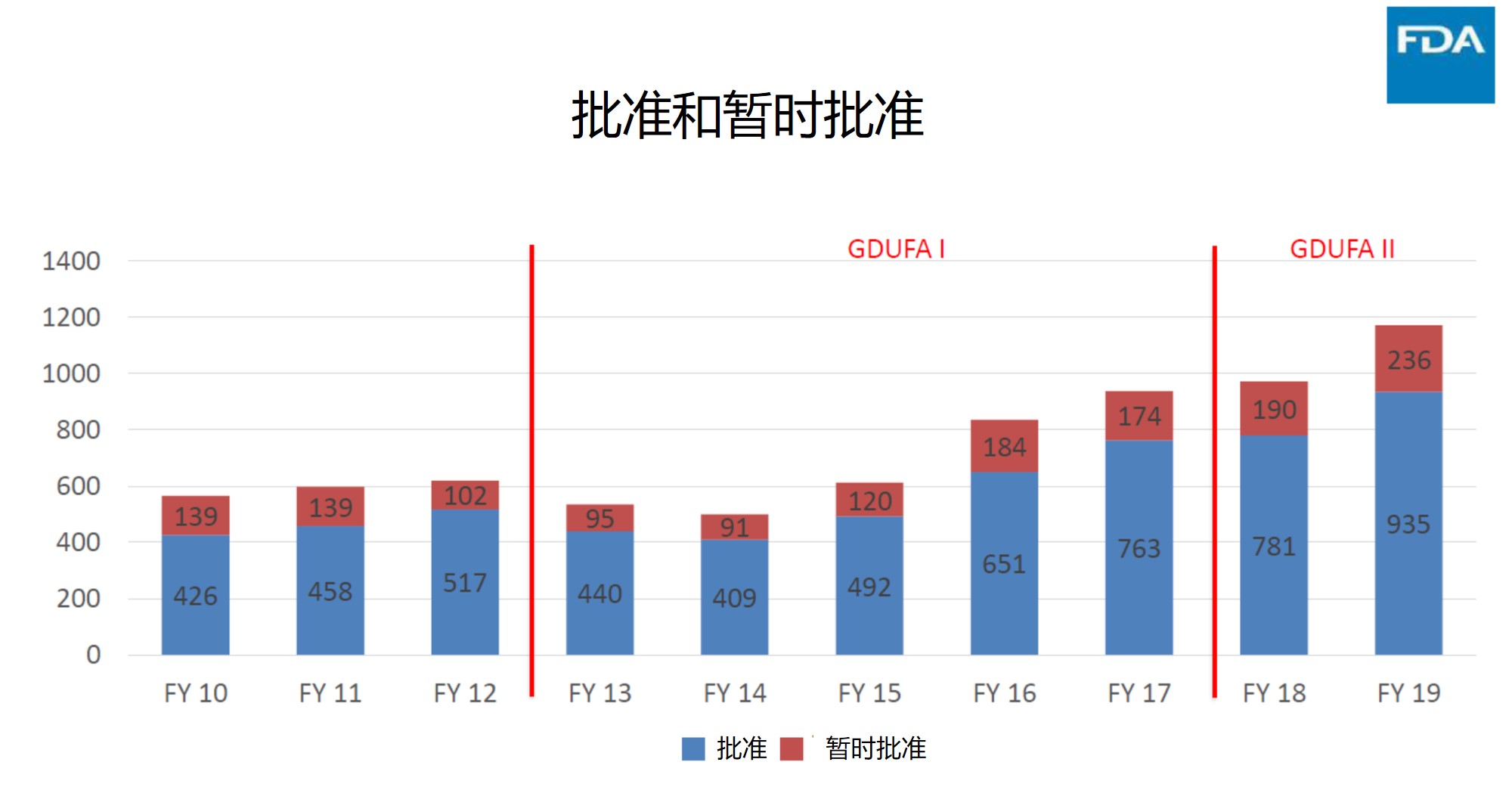
批准与暂时批准
ANDA 批准和暂时批准是代表 ANDA 审批过程最终价值的真实指标,因为如果没有这些最终措施,业界和公众将无法以可负担得起的价格获得高质量的仿制药品。下图显示了 GDUFA 在使仿制药更方便可及方面的成功。在图中可以看到破纪录的审批绩效始于 2015 财年,一直持续到 2019 财年。但按照当前 2020 财年前 6 个月的批准率来看,2020 财年的批准数量可能会大大降低,估计完全批准将达到约 680 件,暂时批准将达到 155 件左右,总的批准绩效估计与 2016 财年相近。
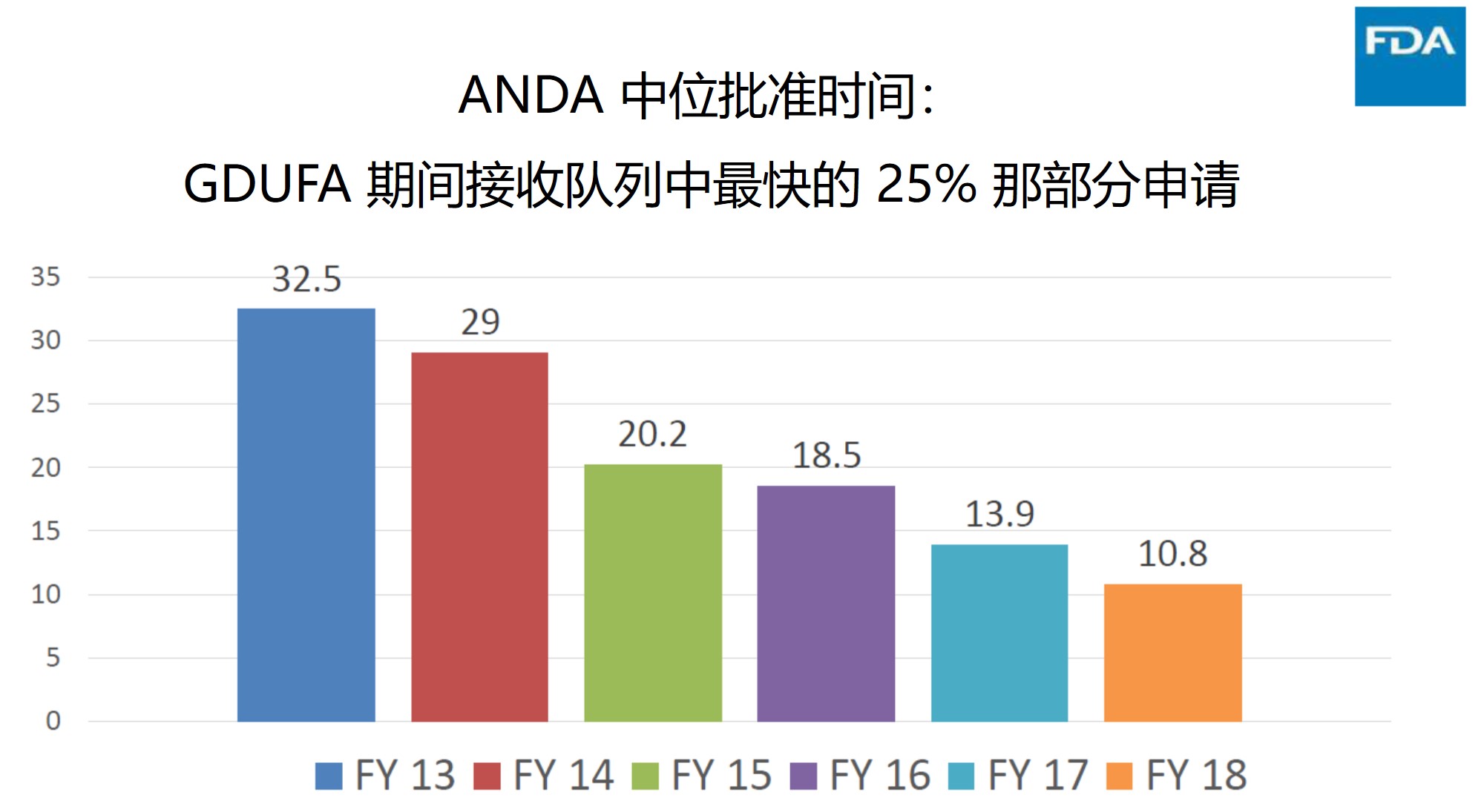
中位批准时间
关于中位批准时间的图表有些混乱,即使在听完演讲后,仍难抓住重点。中位批准时间一直是人们关注的指标,下图似乎表明,在 GDUFA 期间,批准时间的中位数显著下降,并将继续下降。随着申请程序质量的提高,人们期望批准时间的中位数会降低,但是此图显示的是接收队列中最快的 25%,因此,对于企业而言,如果你提交的文件质量很高,可以期望更快的批准时间,但是 GDUFA 每年整个队列的审批时间中位数呢?我们知道,如果查看所有 ANDA 的批准时间而不论其队列年,则中位批准时间将大大增加。如果严格按照队列年份进行统计,那么我们需要相当长的时间才能获得真实数字,因为 2018 财年队列中还有相当数量的申请尚未获得批准,因此随着这些申请陆续在系统中获得审评,中位数将会进一步增加。
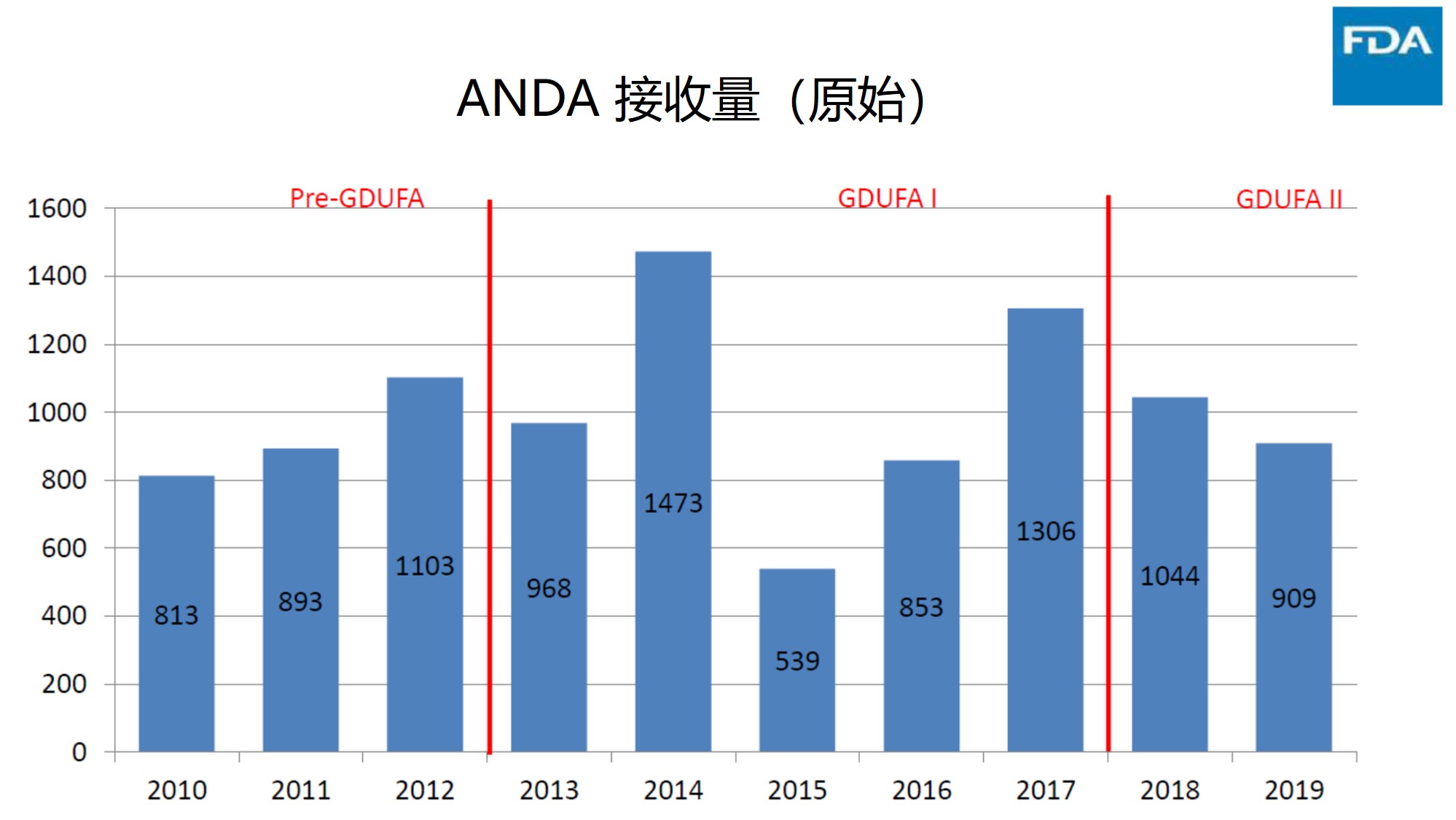
接收量下降
下图显示了在 2014 财年和 2017 财年创下新高之后,原始 ANDA 接收量开始下降。根据现有数据,2020 财年看起来可能会比 2019 财年高一些,以目前的接收率估计全年接收量将在 940 件左右。
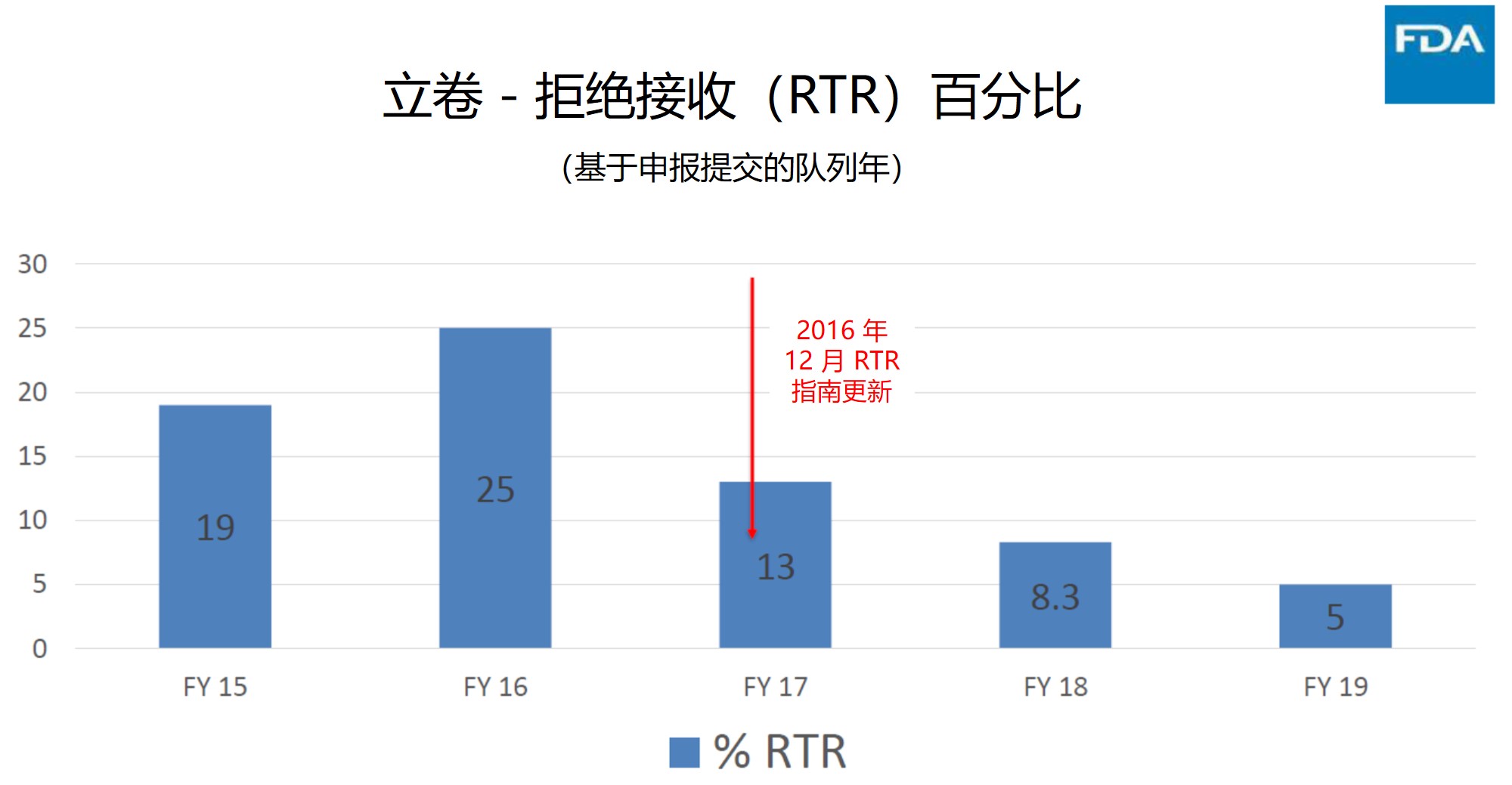
拒绝接收(RTR)
拒绝接收(Refuse to Receive,)一图表明了企业的理解有了显著改善,并且 ANDA 提交的质量也得到改善,因为 RTR 的百分比已降至 5% 的历史最低点。GDUFA I 期间曾专注于解决 RTR 问题,因为其浪费了FDA 和企业资源。2016 年 12 月 RTR 更新指南的发布为行业改善 ANDA 质量提供了绝佳的路线图。
Sherwood 对于立卷和申报给出了一些重点建议,关于立卷:
提交完整的申请 — 避免拒绝接收
使用 ANDA 立卷检查清单
完整填写并签署(并在适当情况下复签)FDA 356(h) 表
提交 ANDA 之前,先在 FDA “可供参考清单”中确认相关的 II 类 API 药物主文件(DMF )
全面并及时的回复信息请求(IR)
在 IR 回复中引用问题
在更正经沟通的电子通用技术文件(eCTD )缺陷时:
就已发现的缺陷遍查所有文件
在需要的地方更正
不要重复原始申报中的信息
使用正确的操作属性(删除/替换)
通过esub@fda.hhs.gov联系CDER ESUB 以在规定的时间内解决问题
将信息放置在 ANDA 层次结构的适当部分
关于申报方面,他指出,
使用清晰的封面函
申报类型
确定优先级(原始和增补)并说明理由
帮助确定潜在的咨询
帮助将你的申报定向到对应的审评组
在引用申报前设施通信(PFC)时
提交完整且清晰的 PFC 及后续 ANDA
保持 PFC 和后续 ANDA 一致
在 PFC 和后续 ANDA 中明确注明优先级要求,并说明理由
如果属于 ANDA 申报前计划的一部分,请在 ANDA 中明确注明
他尤其强调,应考虑审评员的需求,是否想让审评员搜索或评估数据?考虑审评员想看哪些信息。草率的申请是没有办法向审评员传达高质量的研发的。在呈现数据方面,审评员喜欢分析数据,应确保所有数完整呈现,使用清晰的标签和图表,使数据易于阅读和比较,确保数据支持申请中讲述的整个故事,使用数据证明你了解你的产品。整个 ANDA 的编写就是在讲故事,因此如何讲述这个故事,让别人能够了解你对产品的认识很重要。
他一再重申申请人 要了解自己的产品,并在申请中体现出这种了解。关于申请应该“说你所做。做你所说。证明并改进。”另外,关于合同商(包括 DMF 等),应选用负责人的合同商,并与合同商有着积极开放的双向交流,了解并在 ANDA 中列出所有设施 (包括生产、生物分析、临床)。Sherwood 强调,25% 的 ANDA 引用的 DMF 中有隐藏设施。【“隐藏设施”成 FDA 仿制药申请受阻的主要缺陷之一 2019/11/20】
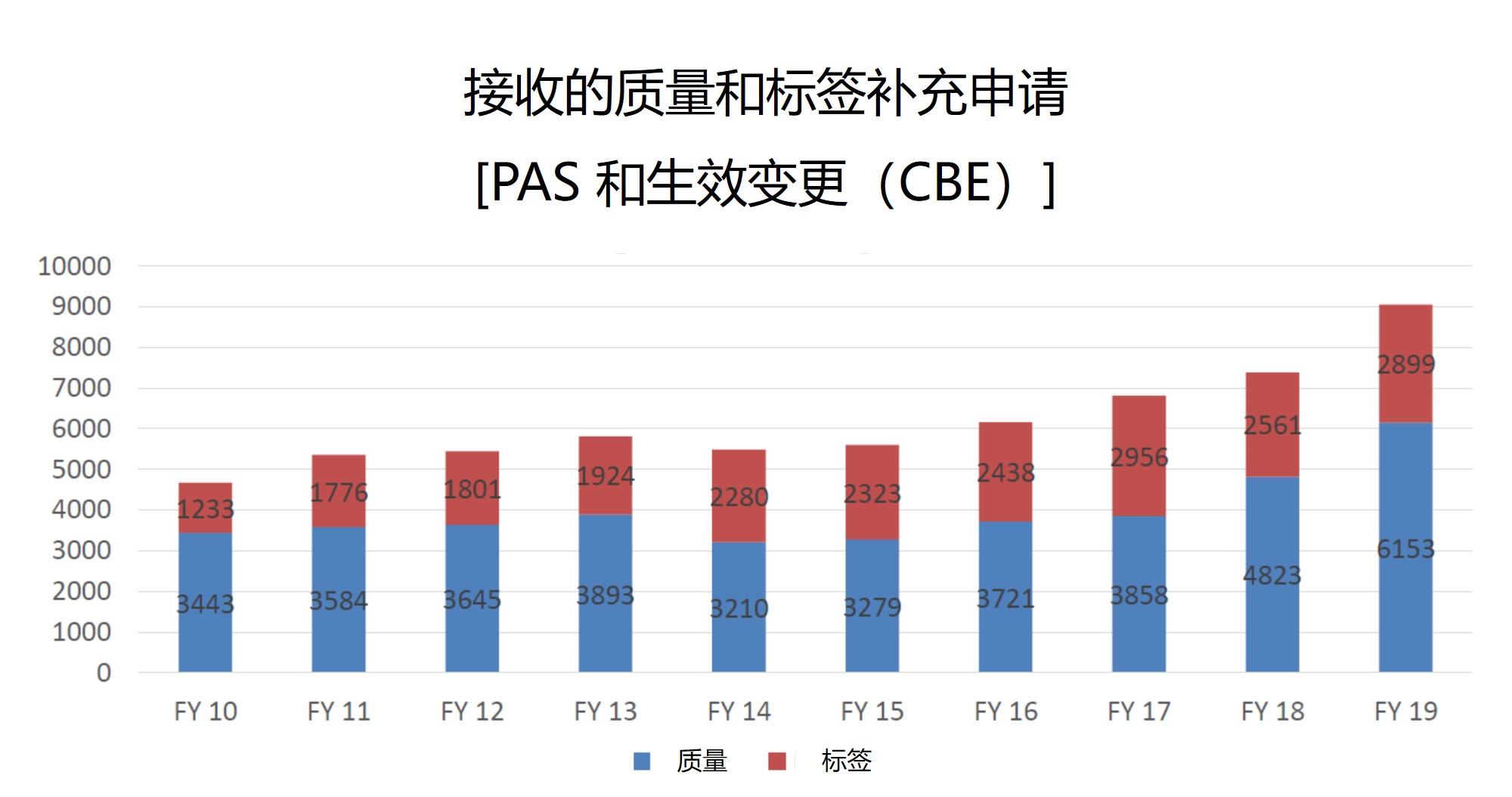
补充申请增加
尽管 ANDA 的提交量似乎在下降,但 ANDA 补充申请 的数量却在增加。众所周知,围绕 ANDA 的活动不会随着批准而中止。批准后,申请人可以寻求各种需要补充提交的批准后变更,这些数字有些惊人,代表着工作量从对原始申报材料的审评转移到对补充申请的审评。
关于仿制药论坛的更多内容,识林将会在此后继续分主题报道。
作者:识林-椒® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
解读法规指南:FDA_Sample_formats_for_Form_FDA_356h_2011
适用岗位必读:
临床研究协调员(CRC):确保临床试验报告的格式符合FDA要求。 注册专员(RA):熟悉FDA 356h表格的填写规范,指导临床试验报告的提交。 药物警戒专员(PV):了解临床试验安全性报告的格式,以便及时准确地报告。 工作建议:
CRC:在临床试验过程中,记录并整理所有必要的数据,确保报告的完整性和准确性。 RA:审查临床试验报告,确保其格式和内容符合FDA 356h表格的要求。 PV:熟悉安全性报告的格式,确保在规定时间内提交给监管机构。 文件适用范围:
文件要点总结:
表格格式要求: 明确了FDA 356h表格的格式要求,包括临床试验的基本信息、安全性信息和研究结果的报告格式。安全性信息报告: 强调了临床试验中观察到的不良事件和严重不良事件的报告要求,要求详细记录事件的严重程度、持续时间和处理措施。研究结果报告: 规定了临床试验结果的报告方式,包括疗效评估、安全性评估和统计分析结果。数据完整性: 要求报告的数据必须准确无误,不得有任何遗漏或错误。提交时间要求: 明确了临床试验报告的提交时间,要求在规定时间内提交给FDA。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA(质量保证) :应全面理解ICH Q12指南,确保质量体系与监管要求一致,指导产品生命周期管理。注册部门 :需熟悉ICH Q12指南,以便在药品注册过程中有效应用,确保申报材料符合监管要求。研发部门 :应了解ICH Q12指南中关于药品开发阶段的技术和监管考虑,以促进创新和持续改进。生产部门 :需掌握ICH Q12指南,特别是在已建立条件(ECs)和变更管理协议(PACMP)方面的要求。文件适用范围:
文件要点总结:
变更管理框架 :提供了一个框架,以更可预测和高效的方式管理批准后CMC变更。已建立条件(ECs) :明确了MAH与监管机构之间的共识,规定了确保产品质量的要素,以及变更时需要进行监管沟通的条件。变更管理协议(PACMP) :提供了一种监管工具,允许MAH与监管机构就变更所需的信息和监管提交的类型达成预先协议。产品生命周期管理(PLCM)文件 :作为ECs和变更报告类别的中央存储库,捕捉商业阶段产品如何被管理。药品质量体系(PQS)与变更管理 :强调了PQS在管理供应链和产品生命周期中的变更管理的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。