首页
>
资讯
>
FDA 临床药理学办公室2024年报,公开审评总结及大量重要文献
出自识林
FDA 临床药理学办公室2024年报,公开审评总结及大量重要文献
2025-07-03
6月13日,FDA发布了《临床药理学办公室2024年度报告》 。该报告详细展示了临床药理学办公室(Office of Clinical Pharmacology,OCP)在过去一年中在药物监管、临床药理学 研究以及推动创新药物开发等方面的工作成果与进展。对于我国监管与药企,OCP的大量研究文献具有独特的参考价值。
OCP是FDA药品评价与研究中心(Center for Drug Evaluation and Research,CDER)转化科学办公室(Office of Translational Sciences,OTS)下的一个多学科办公室。该办公室拥有超过270名专业人员,包括药理学家、药师、生物学家、化学家、医师等。OCP致力于以患者为中心 ,将临床药理学原理应用于确保人用药品和生物制品 的安全性、有效性和最佳使用。
参与审评:基于问题的审评方法
OCP的监管审评综合了所有相关临床药理学知识领域的信息,包括药物处置、药理学 和生物标志物 、定量方法、药物安全、药物治疗学(pharmacotherapy)和临床试验方法,以辅助监管决策。
OCP采用以患者为中心、基于问题的策略,评估新药临床试验申请(Investigational New Drug,IND )、新药申请(New Drug Application,NDA )和生物制品许可申请(Biologics License Application,BLA )的申报信息,以解决剂量选择和优化、治疗个体化以及获益/风险平衡等问题。
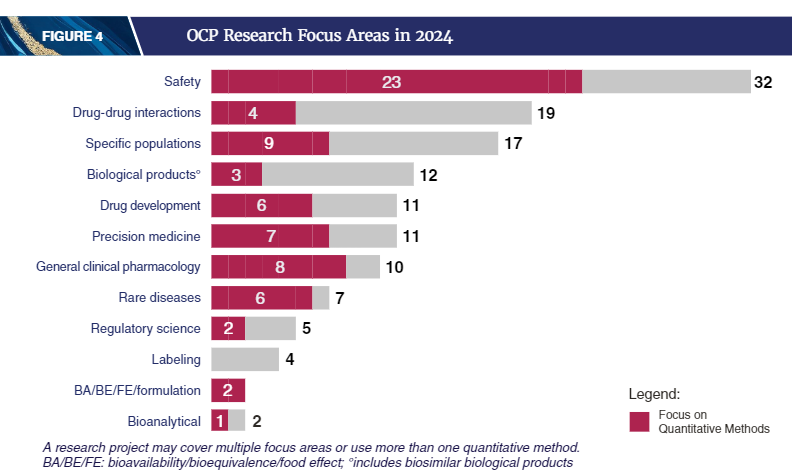
2024年,OCP共进行了超过5400次IND审评,举办了超过3300次药物开发会议,并参与批准了50种新型药品和生物制品。
OCP采用了一种问题导向的方法来评估药物。其评估的关键问题包括:
现有的临床药理学信息在多大程度上提供了有效性的确证性证据和其他证据?
提出的给药方案是否适用于目标适应症 的普遍患者群体?
针对亚人群,是否需要基于内在因素的替代给药方案或管理策略?
是否存在具有临床相关性的食物-药物或药物-药物相互作用,以及适当的管理策略是什么?
定量医学:适应症外推,药物相互作用 以及剂量优化
报告中大量篇幅阐述定量医学(Quantitative Medicine,QM)方法的应用,包括药效学生物标志物的剂量-反应和暴露-反应分析以及药代动力学建模和模拟,在2024年为众多FDA批准的药物提供了有效性的确证性证据和其他证据。
在特定情况下,QM可以将疗效从研究充分的人群外推到在疗效试验中研究不足的人群。2024年,OCP审评人员利用QM方法,成功将9种新分子实体的疗效外推至青少年、儿童、婴儿以及体重至少40公斤的患者(无论其年龄大小),其中7种用于罕见病治疗。
为提交给FDA审批的药物优化给药方案是多学科OCP审评团队的核心职能之一。2024年,OCP工作人员使用多种QM方法,将剂量-反应和暴露-反应分析用于安全性和有效性评估,有助于确定普遍人群中最安全、最有效的剂量。OCP根据年龄、体重、肾功能和肝功能、同时进食和用药以及具有特定基因突变的患者亚群,确定更安全、更有效的剂量。群体药代动力学模型用于识别潜在的可变药物暴露来源,并能够进行临床试验之外的用药场景的药代动力学 模拟,包括不同的药物负荷剂量和给药方案。OCP审评团队还通过发布上市后研究要求,以评估肾功能和肝功能受损患者的安全性和有效性,探索额外的给药方案,并解决临床研究中的代表性不足问题,从而帮助确保药物批准后用药的持续优化。
写指南,做研究,发布文献
2024年,OCP支持FDA共发布了八份临床药理学指南,为药物相互作用、抗体偶联药物和寡核苷酸的临床药理学研究、生物等效性研究、肿瘤药物剂量优化、质量平衡以及肾功能受损的影响等主题提供了建议。监管科学研究方面,2024年OCP的研究组合包括103个项目,并在多个重点领域撰写发布了120篇学术出版物(报告中公布了标题和超链接)。
在国际层面,OCP在ICH的框架下完成了两项指南的最终制定,即M12《药物相互作用研究》 和M13A《口服固体速释制剂的生物等效性》 ,简化全球药物开发流程。OCP代表也是ICH M15《模型引导药物研发的一般原则》 工作组的领导者。
在正在进行的研究中,OCP采用体外系统、建模方法和临床研究来表征药物相互作用的机制(包括罕见药物 代谢酶变异的影响)、更好地预测基于转运体的相互作用,并阐明常见药物组合(如镇静精神药物、血清素再摄取抑制剂和阿片 类药物)的联合药效学效应。机制性药效学模型使研究人员能够模拟一系列过量场景,这些模型的预测将为推荐足够的阿片类拮抗剂剂量以逆转阿片类药物过量毒性提供更多的科学证据。生理药代动力学(Physiologically Based Pharmacokinetic,PBPK)建模技术被用于推进母婴健康研究,预测妊娠期间使用的药物的胎儿暴露。还探索了定量系统药理学(Quantitative Systems Pharmacology,QSP)模型用于儿童发育安全预测。
OCP还研究了一系列药效学生物标志物,以评估其在风险缓解、精准医学、肿瘤学和罕见疾病中的效用,同时利用人工智能 /机器学习技术探索临床试验富集策略和识别免疫原性风险因素。此外,OCP的研究活动为心律失常风险评估和心脏安全终点、人用药中亚硝胺 杂质 控制以及治疗性蛋白的免疫原性 评估等领域的监管最佳实践和全球协调提供了信息。
识林-实木
识林® 版权所有,未经许可不得转载
适用岗位及工作建议:
临床研究员(CR) :必读。在设计临床试验时,考虑药物相互作用对试验结果的影响。药品注册专员(RA) :必读。确保注册文件中包含药物相互作用研究的相关数据和分析。药物安全专员(PV) :必读。监控药物上市后的药物相互作用报告,并进行风险评估。适用范围:
文件要点总结:
研究设计 :强调了药物相互作用研究在临床试验设计中的重要性,要求对潜在的相互作用进行评估。数据要求 :明确了在药品注册过程中,需要提交的药物相互作用研究数据和分析结果。风险管理 :特别指出了药物相互作用可能导致的风险,并规定了相应的风险管理措施。监管要求 :规定了监管机构对药物相互作用研究的审核标准和要求。国际协调 :鼓励在全球范围内协调药物相互作用研究的方法和标准,以促进国际药品注册的一致性。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
生物等效性研究人员:深入理解并应用本指南进行生物等效性研究设计和数据分析,确保研究的科学性和合规性。 临床研究部门:在设计临床试验时,考虑本指南对受试者选择、研究设计、样本量计算等方面的要求。 质量管理(QA):确保生物等效性研究的执行和记录符合本指南的规定。 注册部门:在药品注册过程中,依据本指南准备和提交生物等效性研究资料。 文件适用范围:
要点总结:
研究目的与背景 :明确了生物等效性研究的目的,即通过药代动力学终点评估口服固体制剂的生物等效性,并强调了数据完整性的重要性。研究设计原则 :推荐使用随机、单剂量、交叉研究设计,并详细说明了研究人群选择、样本量计算、研究设计和条件等关键要素。数据与统计分析 :强调了非重复研究设计的数据呈现和统计分析方法,包括生物等效性分析人群的考虑、数据呈现、统计分析的一般考虑等。特殊主题 :讨论了内源性化合物、其他速释剂型(如口腔崩解片、咀嚼片、口服悬浮液)、固定剂量组合和pH依赖性等特殊主题。文档记录 :要求生物等效性研究报告应包含完整的研究协议、执行和评估记录,并符合ICH E3的格式。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
研发(R&D) :必读。负责模型引导药物研发(MIDD)的规划和实施,确保模型的适用性和评估。注册(Reg) :必读。负责与监管机构沟通,提交MIDD证据,并根据监管要求调整MIDD计划。临床(Clin) :必读。参与MIDD证据的生成,以支持临床决策和药物开发。工作建议:
研发(R&D) :在规划MIDD时,明确模型的目标问题(Question of Interest),并根据模型风险和影响评估结果调整模型评估要求。注册(Reg) :在与监管机构的互动中,使用评估表(assessment table)作为沟通工具,提高透明度,并在规划阶段理解MIDD。临床(Clin) :在临床研究中,利用MIDD证据支持决策,特别是在模型风险和影响较高时,需与监管机构进行早期对齐。适用范围:
文件要点总结:
MIDD证据评估框架: 明确了MIDD证据评估的关键要素,包括目标问题、使用背景、模型影响、错误决策后果、模型风险和模型影响。模型评估: 概述了模型评估的元素,包括验证、验证和适用性评估,并提供了一般性建议。MIDD规划和提交: 提供了模型分析计划(MAP)和模型分析报告(MAR)的建议,以及监管互动和提交的文件要求。监管互动: 鼓励在MIDD规划和证据提交阶段与监管机构进行早期和多学科的互动,特别是在模型风险和/或模型影响预期较高时。技术标准和模型评估: 强调了模型评估应遵循当前接受的标准和/或科学实践,并与模型风险相称。以上仅为部分要点,请阅读原文,深入理解监管要求。