首页
>
资讯
>
FDA召开2022年仿制药论坛:ANDA常见缺陷,数据可靠性,KASA,质量文化等
出自识林
FDA召开2022年仿制药论坛:ANDA常见缺陷,数据可靠性,KASA,质量文化等
2022-05-12
4月26日至27日,FDA 召开了2022年仿制药论坛(Generic Drugs Forum,GDF) ,此次会议通过提供实用建议、介绍真实案例研究以及深入分析简化新药申请(ANDA) 评估过程,聚焦仿制药 的现状,讨论了许多热门话题,如 pre-ANDA 计划、仿制药指标、上市后安全、批准前检查 和全球仿制药事务等等。
下文简述了会上讨论的 ANDA 中的常见缺陷 以及与数据可靠性 相关的问题,完整的会议视频和 PPT 可登录识林观看(视频列表请见文末)。
仿制药论坛2022-仿制药的现状
{{#video:v300c2372-8113-415a-8232-b437da73dec5}}
ANDA 中的常见缺陷
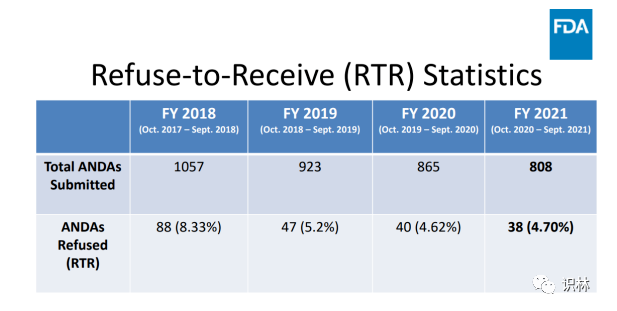
与往年相比,2021财年 FDA 通过其拒绝接收 (refuse-to-receive,RTR)机制拒绝的 ANDA 数量较少,这归因于行业透明度的提高、更好的申请实践以及与申请人 沟通的加强。
FDA 的努力,识林曾有多次报道,如相关资讯【FDA 发布 ANDA 缓释片生产缺陷评估报告,帮助仿制药申请人规避缺陷提高首轮批准率】 。
论坛第二天,评审专家 Peter Enos 讨论了行业如何改进文件的提交以避免 RTR 。FDA 数据显示,在2021财年,808份 ANDA 申请中有4.7%被拒收,低于2018财年的1057份申请中8.33%的拒收率。2021财年引起 RTR 的主要缺陷 为稳定性数据不足以及与参比制剂 (reference-listed drug,RLD)定性/定量(Q1/Q2)一致性的研究不充分。其他问题包括溶出度研究不充分、杂质鉴定不充分、生物等效性 研究不充分,以及使用某些辅料 的理由不充分。
为了确保稳定性数据的可接受性,Enos 表示“每个规格必须提供至少三个试验批次,来自两个批次的 API ,且数据应包括需要三个时间点的至少六个月的加速 和长期稳定性 研究。”如果在加速研究的任何时间点出现稳定性超标,则应进行中间条件稳定性研究(Intermediate studies)。
为了避免定性/定量(Q1/Q2)比较的不充分,申办方应确保非肠道药品(注射剂 )包含与 RLD 相同的辅料,浓度也相同。某些辅料(即防腐剂 、缓冲剂、抗氧剂)允许与 RLD 有差异,但需标注不同之处,阐述理由,并研究证明上述不同不影响所申请产品的安全性和有效性,但眼用药不得使用与 RLD 不同的辅料。
为确保溶出度研究充分,建议申办者参考 FDA 的特定产品指南 (product-specific guidances,PSG,最新的 PSG 指南于2022年2月发布),其中包括溶出度研究建议和其他有关开发特定仿制药 的建议。溶出度研究应包括受试产品和 RLD 的每种规格至少12个剂量单位。
此外,电子通用技术文档 (eCTD) 中的提交内容应清晰易读,页面导航应正确,提交内容应包含足够的描述性书签或超链接。
ANDA 中的数据可靠性问题
在为期两天的仿制药 论坛上有七个关于数据可靠性 (Data integrity,DI)的单独演讲,占据了论坛第一天几乎一半的时间,也提示了 DI 问题在行业中仍然存在并且十分重要,而 FDA 再次将重点转向识别和解决 DI 带来的问题。
仿制药办公室(OGD)副主任 Nilufer Tampal 指出,质量文化 (Culture of Quality)薄弱的公司更容易提交存在数据可靠性问题的 ANDA。她在演讲中强调了与 DI 相关的各种问题,涵盖生物等效性 研究、药理学/毒理学研究、临床和分析研究以及 ANDA 中的 DI,并讨论了解决问题的方法以及对 DI 的一些常见误解。
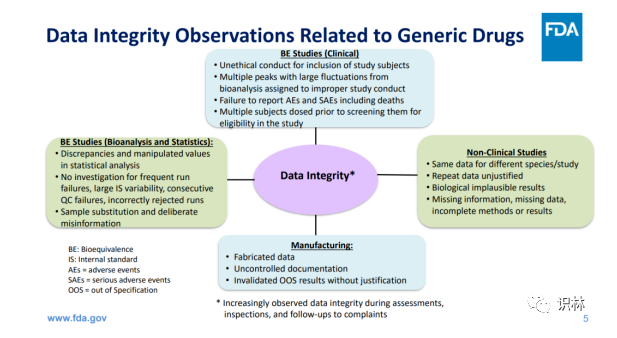
仿制药申请和开发过程中数据可靠性问题可以分为以下四类:
上图内容翻译如下:
生物等效性研究/临床:
招募研究受试者的非伦理行为(Unethical conduct)
未报告不良事件 (AEs)和严重不良事件(SAEs),包括死亡
BE 研究(生物分析和统计):
统计分析中的差异和操纵值(manipulated values)
没有针对频繁运行故障、内标(Internal standard,IS)可变性、连续 QC 故障、不合理的剔除数据(incorrectly rejected runs)进行调查
非临床研究 :
生产:
无正当理由的无效超标 (Out of specification, OOS)结果
对数据可靠性的误解
关于数据可靠性违规的常见误解,一是数据可靠性问题只会出现在质量控制实验室 ,这是错误的,因为这些问题可能发生在公司的任何地方;另一个误解是领导层对 DI 问题“不负责任、不可能知道”,这也是错误的,解雇一名应对数据可靠性 违规负责的员工不能解决问题。
质量文化有“缺陷”的公司更容易受到数据可靠性问题的影响,拥有强大质量文化的公司能够更好地防止 DI 问题的发生。强大的企业质量文化是指“领导层致力于促进支持数据可靠性和产品质量的工作环境,并制定了有关行为标准的详细政策”。公司需要通过持续培训不断强化质量文化,领导层应强调数据质量是每个人的责任,这一点很重要。强大的质量文化鼓励公开透明的沟通,尤其是在错误报告和偏差 方面。对质量文化的强调,识林曾有评论文章:【质量对话:质量文化的两项标志】
最后,Tampal概括了数据可靠性审查的关键要点:
FDA 期望在ANDA 中提交的所有数据都是可靠和准确的
申请人 有责任确保所有其他来源研究活动的数据可靠性
领导层应积极投资于工具和政策,以增强数据可靠性
质量管理体系 (Quality Management System, QMS)和企业质量文化不是一劳永逸的事情
下表为此次论坛的所有演讲与讨论,视频内容和相关PPT可至识林查看。
第一天
1
主旨演讲 - OGD 和 OPQ
Keynote - OGD & OPQ
2
知识辅助评价和结构化申请 (KASA):药品评价
Knowledge-Aided Assessment and Structured Application (KASA): DRUG PRODUCT ASSESSMENT
3
通过 KASA 实现制造评估现代化
Modernizing Manufacturing Assessment through KASA
4
KASA 在生物药剂学评估中的应用
Use of Knowledge-Aided Assessment and Structured Application (KASA) in Biopharmaceutics Assessment
5
综合质量评估 - 药品
Integrated Quality Assessment - Drug Product
6
问题与小组讨论
Questions & Panel Discussion
7
概述:仿制药 计划年度统计
Overview: Generic Drug Program Annual Statistics
8
公共统计数据 - 它们是什么?它们意味着什么?
Public Stats - What Are They and What Do They Mean
9
公共统计数据及其含义 - 仿制药政策办公室
Public Stats and What They Mean - Office of Generic Drug Policy
10
ANDA 计划公共统计数据及其含义 - 药品质量办公室
ANDA Program Public Stats and What They Mean - Office of Pharmaceutical Quality
11
问题与小组讨论
Questions & Panel Discussion
12
质量文化
Culture of Quality
13
申请提交中的数据可靠性 和数据质量
Data Integrity and Data Quality in Application Submissions
14
BA/BE临床现场检查的数据可靠性问题:案例研究和OSIS评估
Data Integrity Issues from BA/BE Clinical Site Inspections: Case Studies and OSIS Evaluation
15
分析数据可靠性 - 透过现象看本质
Analytical Data Integrity - Looking Beyond the Obvious
16
药理学 毒理学 研究中的数据可靠性
Data Integrity in Pharmacology Toxicology Studies
17
数据可靠性在药物应用中的作用
Role of Data Integrity in Drug Applications
18
问题与小组讨论
Questions & Panel Discussion
第二天
1
FDA 关于人用药中亚硝胺 杂质的控制概述
FDA Overview Control of Nitrosamine Impurities in Human Drugs
2
液体制剂常见制造相关缺陷
Common Manufacturing Related Deficiencies for Liquid Drug Products
3
问题与小组讨论
Questions & Panel Discussion
4
仿制药开发和全球不同的监管法规
Generic Drug Development and Globally Divergent Regulations
5
FDA 产品特定指南计划概述
FDA Product-Specific Guidance Program Overview
6
使用前瞻性研究支持产品特定指南(PSG)开发的方法
Approaches Using Proactive Research in Support of Product-Specific Guidance (PSG) Development
7
问题与小组讨论
Questions & Panel Discussion
8
仿制药办公室对研究性新药申请 (Bio-IND) 的审评
Review of Investigational New Drug Applications (Bio-INDs) by the Office of Generic Drugs
9
Pre-ANDA 会议计划概述
Overview of Pre-ANDA Meeting Program
10
问题与小组讨论
Questions & Panel Discussion
11
与 FDA 沟通的最佳实践和策略
Best Practices and Strategies for Communication with FDA
12
立卷审查部门:ANDA和受控函 提交的最佳实践
Division of Filing Review: Best Practices for ANDAs and Controlled Correspondence Submissions
13
问题与小组讨论
Questions & Panel Discussion
14
上市前和上市后仿制药安全项目管理
Project Management of Premarket and Postmarket Generic Drug Safety
15
在 ANDA 中进行可比性 分析的最佳实践
Best Practices for Conducting Comparative Analyses in ANDAs
16
问题与小组讨论
Questions & Panel Discussion
17
在 COVID-19 大流行期间使用替代工具进行检查
Use of Alternate Tools for Inspections during the COVID-19 Pandemic
18
评估药品质量体系 的有效性:质量监督办公室的视角
Assessing the Effectiveness of a Pharmaceutical Quality System: Office of Quality Surveillance Perspective
19
OPQ 政策更新:ICH Q12
OPQ Policy Update: ICH Q12
20
问题与小组讨论
Questions & Panel Discussion
作者:识林-红木
识林® 版权所有,未经许可不得转载。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA:负责确保实验室操作符合质量控制要求,监督取样、留样、检验等流程。 研发:在设计质量标准和分析方法时,需遵循本文规定。 生产:在取样和留样过程中,应遵守本文的详细规定以保证产品质量。 文件适用范围:
文件要点总结:
实验室职责与布局: 明确了质量控制实验室的职责、布局原则和要求,以及人员的组织架构和资质要求。取样与留样管理: 规定了取样过程的控制和留样的定义、量、储存要求及记录。物料和产品检验: 强调了检验要求,包括待检样品核对、检验、记录和报告书的编制。委托检验管理: 阐述了委托检验的原则、应用范围、职责和工作流程。质量标准建立: 详细说明了质量标准的设计与制定、审核与批准流程。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。 适用范围:
文件要点总结:
质量管理体系概述 :明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。产品质量实现要素 :涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。质量保证要素 :包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。质量风险管理 :介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。质量管理系统文件 :规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。