首页
>
资讯
>
ANDA申报流程及材料清单上线了
出自识林
2018-08-29
自片剂工艺验证流程 以来,识林陆续梳理了胶囊剂工艺验证流程 、美国FDA DMF申报流程 、EDQM-CEP申报流程 等场景应用工具,且每个流程的关键步骤都配有参考资料清单,申报资料的编写也做了详细总结。
场景应用工具上线后,用户好评并积极反馈,也有更多专业人士进入识林“植树”,识林将继续推出更多场景案例,欢迎大家通过“设置”—“反馈意见”功能或页面下讨论功能进行反馈、指正、完善案例内容或提出案例需求。
也欢迎更多有志之士加入,分享经验与作品,成为有专业影响力的行业领跑者。详情请点击:【识林求贤 】
本月识林ANDA 申报流程 推出后,识林专家提出了以下几点补充,是容易被注册人员疏忽的内容。
• 除了ESG 递交,还有其他方法?还可以查历史版本?
以下几种递交都可以在CDER direct (https://direct.fda.gov/ ) 完成,并且可以追溯历史版本,非常方便,而ESG只适合于递交。GDUFA Facility Self-Identification (GDUFA 下设施的自我认定 )
• RTR 一年内企业没有任何行动,已递交文件被删除?
基于对21 CFR 314.101 和314.3 的理解,RTR一年内企业没有任何行动,FDA理论上会按主动撤销处理。但是因为当前申请都是eCTD 递交,即sequence 0001、0002,已递交文件都是电子化归档,FDA不会主动删除。那么对于后续的重新递交,需要注意:
• 对制剂生产商的批准前检查(Pre-Approval Inspection,PAI) 和对API 生产商的批准前检查,当前FDA的检查已经启用运营方针(concept of operation)方式,更全面地将人用药药品审评计划与场地评估和检查相结合。 【FDA 加强药品生产检查和监督的新步伐 2017-09-01 】
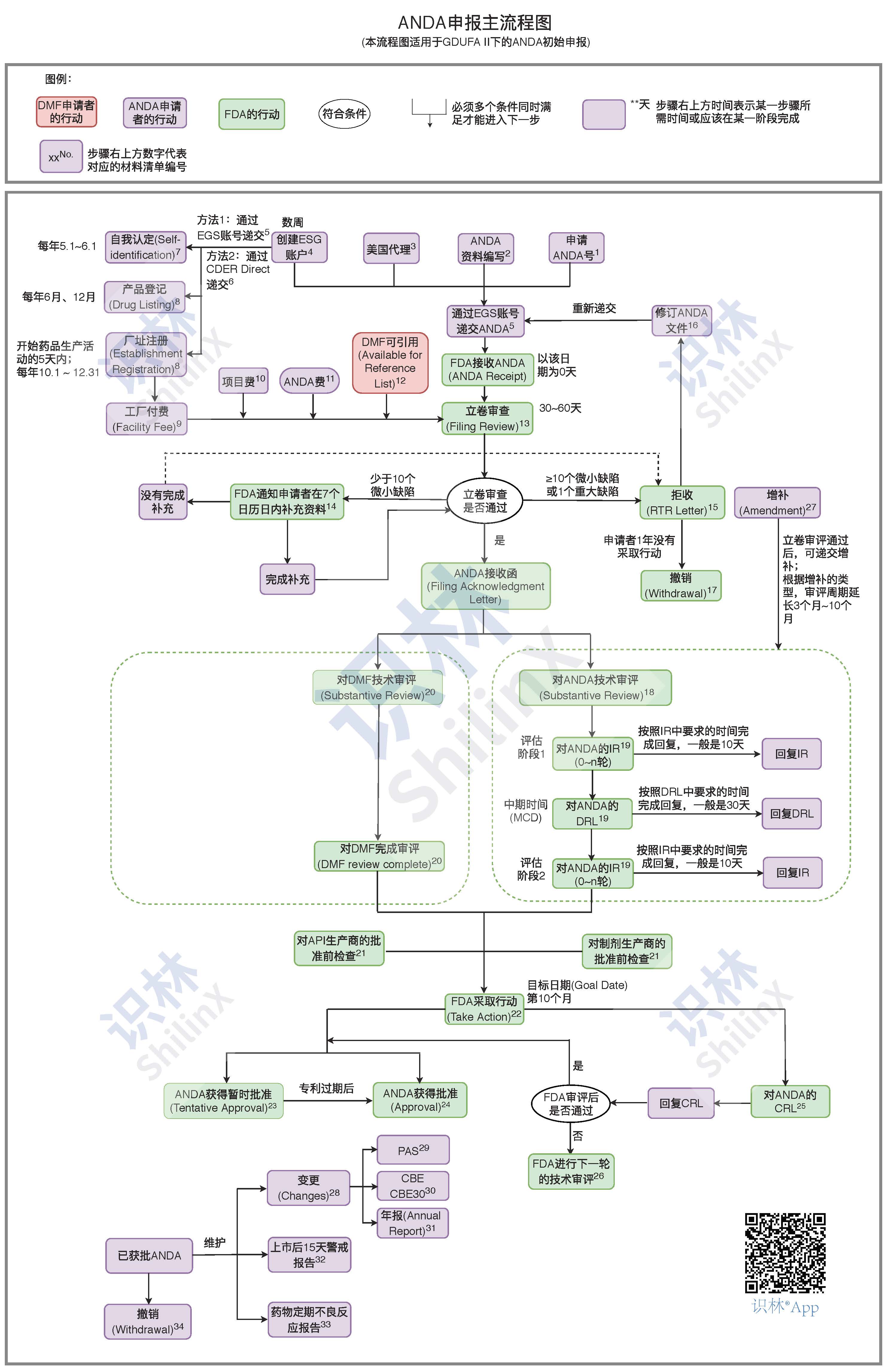
下面的流程图列出了从零开始至上报、审评和后期维护的 ANDA 整个注册周期所涉及到的关键步骤和时限。本流程图适用于2017年10月1日起执行的GDUFA II下的初始递交至FDA的ANDA。识林企业会员可结合材料清单 学习,以流程图为主导,清单为详细了解及操作指导;流程图与清单以编号为桥梁一一对应(识林网页版学习效果更佳。)
注:本流程采用标准、稳妥的现行步骤,其他策略(比如ANDA优先审评 )、非现行法规和指南暂不列入。更多参考资料见识林其他页面。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。