首页
>
资讯
>
起始物料难题新进展 — ICH最新培训资料中文版
出自识林
2018-07-25
2018 年 7 月 20 日,ICH 发布了《Q11 Q&As原料药的起始物料的选择与论证》培训材料 ,ICH 在 2012 年 5 月和 2017 年 8 月就分别发布了 Q11 和 Q11 Q&A 指南,但从 ICH Q7 开始,起始物料 问题就一直是原料药 的开篇难题,这份培训资料探讨了下列问题:
化学合成原料药起始物料的选择
S.2.2 中生产工艺 描述“充分”的重要性
影响原料药杂质谱 的步骤
持续存在于多个步骤中的杂质
在选择起始物料时关于诱变杂质的方法
为了更好的使业界理解在选择起始物料时,应如何应用指南中的一般原则和决策树?这份培训资料提供了三个详细案例,通过数据、实证和细致的图表,手把手的再次向业界阐释了 ICH Q11 和 Q&A 描述的原料药起始物料的选择和论证原则,并强调原料药起始物料的确定应基于科学(数据)和知识(对工艺原理的理解),应考虑 ICH Q11 和 Q&A 部分中所有指导内容。下面是三个案例的概述,完整中文翻译请见此处 。
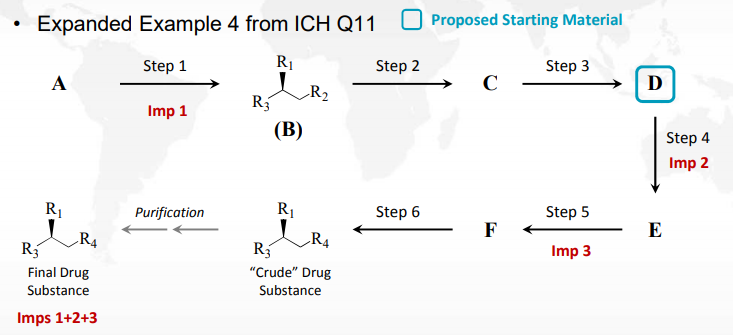
Q11 案例 4 的延伸案例
ICH Q11 建议“影响原料药杂质谱的生产步骤通常应在申请材料的第 3.2.S.2.2 节描述生产工艺。”但是,如 ICH Q11 例 4 所述,当杂质产生于早期并“持续”存在于多个步骤中直至原料药,该原则并不一定适用。正如通常所预期,持续存在杂质是基于它在拟起始物料上游的一个或多个生产步骤中被带入,此时这些步骤不会影响原料药的杂质谱(对于“影响”,见问答 5.7)。
在培训材料中,对 ICH Q11 的案例4进行了延伸,最终原料药中的杂质都是 Imp 1+2+3,Imp 1(化合物B的对映异构体)是 Step 1 中产生的杂质,在 Imp 2 和 Imp 3 的生成步骤不变化的多种情况中,应该如何选择起始物料。图1是杂质生成步骤的情况之一,Imp 2 和 Imp 3 分别在 Step 4 和 Step 5 中生成,在 Step 2 和 Step 3 中没有产生影响原料药杂质谱的杂质,此时选择 D 作为起始物料。(选择起始物料的一般原则、关键考虑点等详见培训材料识林中译版)。如果涉及诱变杂质,还应当在选择起始物料时运用 ICH M7 的原则。
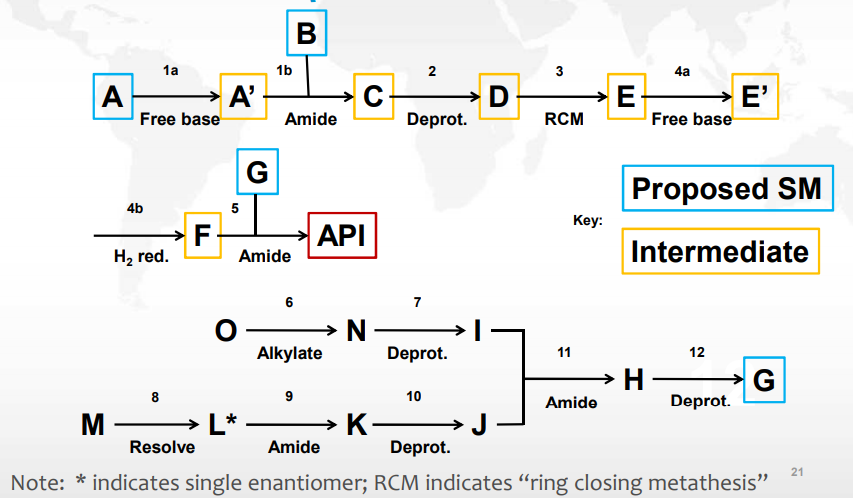
案例研究 1 重新选择起始物料
申请人提出将蓝色框圈出的 A、B、G 作为起始物料,杂质来源如下表中所述。通过分析合成路线、杂质来源及控制策略以及运用 Q11 Q&A 与决策树,得出结论:
步骤 6 和 8 中产生的杂质 - 持续存在于原料药中
I 和 J 之前的步骤不产生其他影响原料药杂质谱的杂质,S.2.2 中不需要包括步骤 6-10
经过这些考虑后,仅有很少的化学转化步骤是要列入 S.2.2 的,因此 G 不能作为起始物料
起始物料 G 现在是作为中间体
申请人将 I 和 J 作为起始物料是可以接受的
重新定义起始物料后,就有了充分的工艺描述
杂质
来源
i
Step 6(持续存在的诱变杂质)
ii
Step 4b
iii
起始物料 A上游的4步以上
iv
步骤5
v
起始物料 B上游的2步以上
vi
起始物料 B上游的2步以上
vii
步骤8(持续存在的对映异构体杂质)
viii
步骤5
ix
步骤5
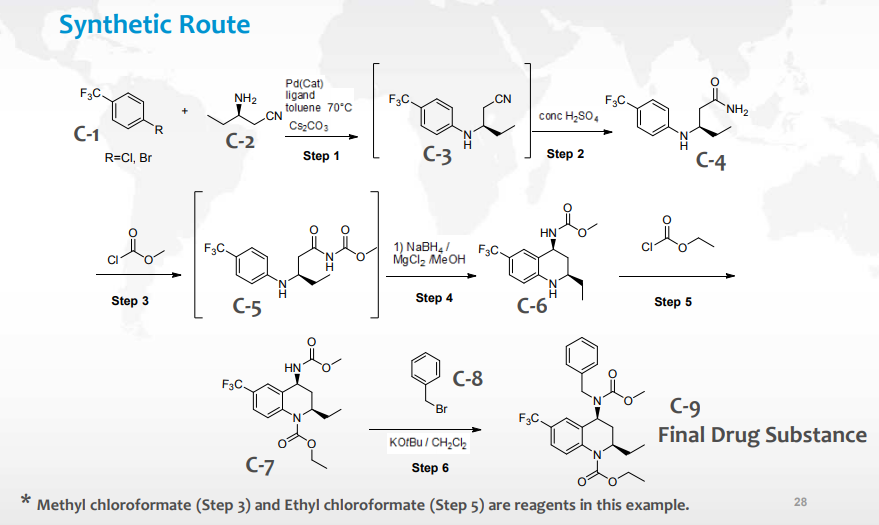
案例研究 2 基于 Sakuramil mock dossier 选择起始物料
案例中分别对 C-9-D1、C-8、C-5、C-7、C-6、C-4 进行细致的分析,判断是否可被作为起始物料。例如 C-8 benzyl bromide(溴化苄)是市售可获得的化学品,同时是在合成的最后步骤中被引入的重要结构片段(不是试剂),由此可判断 C-8 可以作为起始物料;C-5 是不可分离中间体,通常不适合作为起始物料;C-7 的上游步骤会产生影响药物杂质谱的杂质,若选取合成路径后端的中间产物作为起始物料,那么就无法确保生产工艺在充分的 GMP 条件进行,所以 C-7 不可以作为起始物料等。
杂质
申请人为支持自己的论证而描述的考虑事项
C-9-D1
Step 4(C-7中的残留杂质转化为C-9的非对映异构体即C-9-D1)
相关物质
所有非诱变杂质是在商业开发过程中发现的
可诱变杂质
根据每日剂量,对于API,M7的毒理学关注(TTC)阈值为25ppm
C-8
来自Step 6的未反应的拟起始物料C-8是已知的诱变剂
C-6
C-6会影响API
C-3,C-4,C-5
C-3,C-4,C-5不影响API
适用岗位:
QA(质量保证):负责监督和确保药品生产过程符合M7(R1)指南要求,确保基因毒性杂质控制在可接受水平。 R&D(研发):在新药研发过程中,需评估和控制合成过程中产生的基因毒性杂质,确保药物安全性。 临床研究:在临床试验阶段,需根据M7(R1)指南评估药物中的基因毒性杂质,确保受试者安全。 注册:负责药品注册文件的准备和提交,需根据M7(R1)指南提供基因毒性杂质的评估和控制策略。 工作建议:
QA:定期审查生产流程,确保基因毒性杂质的控制措施得到有效实施,并符合M7(R1)指南要求。 R&D:在药物合成和制剂开发阶段,主动识别和评估潜在的基因毒性杂质,采取控制措施以降低风险。 临床研究:在设计临床试验方案时,考虑基因毒性杂质的风险评估,并在试验报告中包含相关数据。 注册:在药品注册文件中明确说明基因毒性杂质的评估结果和控制策略,确保符合监管要求。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议 研发(R&D) :理解原料药开发和生产过程中的关键质量属性(CQAs)及其与药物产品的关系。质量管理(QA) :掌握文件中提到的质量风险管理(QRM)和控制策略,确保生产过程的合规性。注册(Regulatory Affairs) :熟悉CTD格式下原料药生产过程开发和相关信息的提交要求。生产(Production) :了解生产过程描述和过程控制,以及如何在生产过程中应用设计空间。文件适用范围 本文适用于化学药品和生物技术/生物制品的原料药开发与生产。特别针对Common Technical Document (CTD)格式下第3.2.S.2.2 - 3.2.S.2.6节的内容组织。不适用于临床研究阶段的提交内容。发布机构为国际协调会议(ICH),适用于跨国药企和Biotech公司。
文件要点总结 生产过程开发目标 :确立能够持续生产预期质量的原料药的商业生产过程。原料药与药物产品的质量关联 :原料药的预期质量应通过其在药物产品中的使用以及对其物理、化学、生物和微生物特性的理解来确定。生产过程历史与变更管理 :描述原料药生产过程的显著变化,并评估这些变化对原料药质量的潜在影响。控制策略 :基于当前产品和过程理解,确保过程性能和产品质量的计划控制集合。设计空间 :已证明能提供质量保证的输入变量和过程参数的多维组合和交互作用。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位必读建议:
QA(质量保证部门):应全面理解并执行GMP原则,确保所有相关活动符合规定要求。 生产部门:必须遵循文件中关于生产操作、设备维护和清洁、校准以及计算机化系统的规定。 研发部门:在API的初期研发阶段,应考虑GMP原则以确保产品质量。 临床部门:在临床试验阶段使用API时,应遵守特定的GMP实践,确保临床试验材料的质量。 文件适用范围:
文件要点总结:
质量管理体系 :强调所有参与制造的人员应负责质量,建立并实施有效的质量管理体系。人员 :确保足够的合格人员参与API的制造,并进行定期培训。建筑与设施 :建筑设计应便于清洁、维护,并最小化污染风险。生产设备 :设备的设计、建造和维护应确保不影响API的质量,并定期校准。文件和记录 :所有相关文档应按照书面程序准备、审核、批准和分发,确保记录的完整性和可追溯性。材料管理 :建立材料接收、鉴定、隔离、存储、取样、测试和批准或拒绝的书面程序。生产和过程控制 :监控生产步骤的进展并控制过程变异性,确保API的一致性。验证 :包括验证政策、文件、资格认证、过程验证方法和周期性审查已验证系统的程序。变更控制 :建立正式的变更控制系统,评估可能影响中间体或API生产和控制的所有变更。投诉和召回 :记录并调查所有质量相关的投诉,并制定召回程序。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。