首页
>
资讯
>
研究显示 FDA 的首创新药加速审评远超 EMA
出自识林
研究显示 FDA 的首创新药加速审评远超 EMA
2025-04-01
根据最新发表在Health Affairs上的研究《美国和欧洲对首创药的监管存在差异》 (First-in-Class Drugs Experienced Different Regulatory Treatment in The US And Europe),FDA比EMA更倾向于为首创新药提供加速审评和监管灵活性。但这种加速审评模式也带来了更高的不确定性,上市后监督仍需加强。
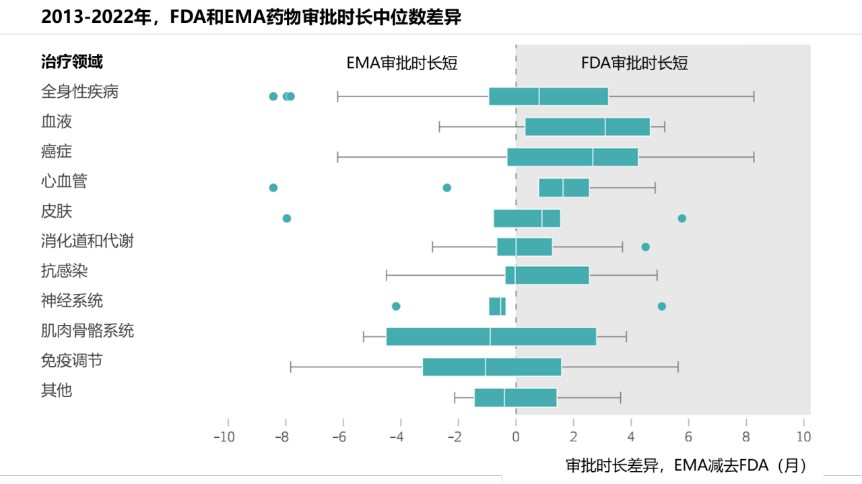
FDA与EMA加速审评研究结果
研究基于2013年至2023年间FDA批准的186种首创新药,以及2013年至2022年间FDA与EMA共同批准的121种药物,评估了FDA和EMA的审评时长、药物是否获得加速审评 、药物相关的治疗领域以及支持药物批准的关键试验的特征。(如下图,翻译供参考)
从审评时长而言,仅由FDA批准的药物的审评时长为8.17个月;而FDA和EMA共同批准的药物,前者的审评时长为8.13个月,后者为10个月。
在FDA批准的186种药物中,150种药物(80.6%)获得了一种或多种加速审评,例如优先审评 (75.2%)、快速通道认定(45.6%)、突破性疗法(40.8%)或加速批准(18.2%)。在FDA和EMA共同批准的121种药物中,83.4%的药物获得了加速审评,而EMA加速审评(如加速评估、附条件批准或特殊情况)的使用率仅为29.7%。
FDA批准的最常见药物类型与癌症相关,其中45种药物仅由FDA批准(占24.1%),33种药物由FDA和EMA共同批准(占27.2%)。在分析仅由FDA批准的185种基于人体临床试验 的首创新药时,研究人员确定了314项关键试验;其中,109种药物(58.9%)仅有一项关键试验,61种(32.9%)使用了替代指标,46种(24.8%)临床试验采用了外部对照,45种(24.3%)没有支持其批准的III期关键试验。例如,天花治疗药物Tpoxx(tecovirimat)仅根据动物规则批准,因为当时无法在人体中进行测试。
加速审评面临的挑战
耶鲁大学医学院医学和公共卫生教授Joseph Ross指出,加速审评虽然有助于药物更快上市,但也带来了更高的不确定性。由于加速审评通常依赖单一临床试验和替代指标,药物在批准时的证据基础可能不够充分。
因此加速审评更像是制药企业与监管机构之间达成的某种“协议”:由于更快地将药物推向市场,制造商需要确认药物的临床获益,而FDA则需要确保制造商完成后续的确认性临床试验。然而,这一协议并未如期实现,部分药物在获得加速批准后,制造商未能按时完成必要的后续研究,导致药物长期缺乏充分的证据支持 。
Ross特别指出,虽然获得加速批准的首创新药在某些方面取得了进展,但对于获得突破性认定的药物来说,情况并不乐观 。这些药物往往缺乏足够的证据来证明其临床获益,给患者和医疗系统带来了潜在风险。
上市前审评和上市后监督
研究人员Jihye Han强调,监管审评应分为两个阶段:上市前审评和上市后监督。在上市前阶段,监管机构应确保药物具有充分的疗效和安全性证据。而在上市后阶段,必须通过严格的监督来确认药物的有效性和安全性。如果药物未能达到预期效果,监管机构应采取进一步措施,如更新安全警告、限制适应症 或撤销批准。
Han指出,药物批准后,剩余的不确定性将由患者和医疗系统承担。为了减轻无效或不安全药物的风险,加强上市后监督至关重要。然而,FDA在撤销加速批准药物方面的能力存在局限性 ,即使上市后试验未能确认药物的有效性和安全性,撤销批准的过程也可能面临阻力。
随着FDA可能加强监管指导和规则制定 ,未来首创新药的审评标准可能会更加严格。社会对药物证据生成的需求也将推动监管机构在加速审评与证据质量之间做出更好的权衡。更严格的上市前审评和更有效的上市后监督才能确保患者获得安全有效的创新药物。
识林-筱筱
识林® 版权所有,未经许可不得转载