|
首页
>
资讯
>
替代仍然不足,三星报告揭示美国生物类似药市场复杂现状
出自识林
替代仍然不足,三星报告揭示美国生物类似药市场复杂现状
2025-05-15
4月25日,科技巨头三星旗下的生物类似药公司Samsung Bioepis发布了第9份生物类似药季度市场报告。
细读报告,一个可能有些意外的数据是,FDA批准的生物类似药中,还有三分之一未能在美国市场上市。生物类似药要做临床可比性研究(在美国还要做可转换研究),开发成本远高于许多小分子化学仿制药。即便如此,已获批药品却因为种种原因无法上市,而那些上市的生物类似药也并未如业界所期望的那样显著拉低用药成本,更未能出现化药仿制药上市带来的所谓“专利悬崖”现象。这一点不仅提示我国生物类似药出海企业提早关注市场挑战,也侧面印证了近来欧美药监合力推动给生物类似药减负、加速上市,从而强化市场竞争的动机所在。
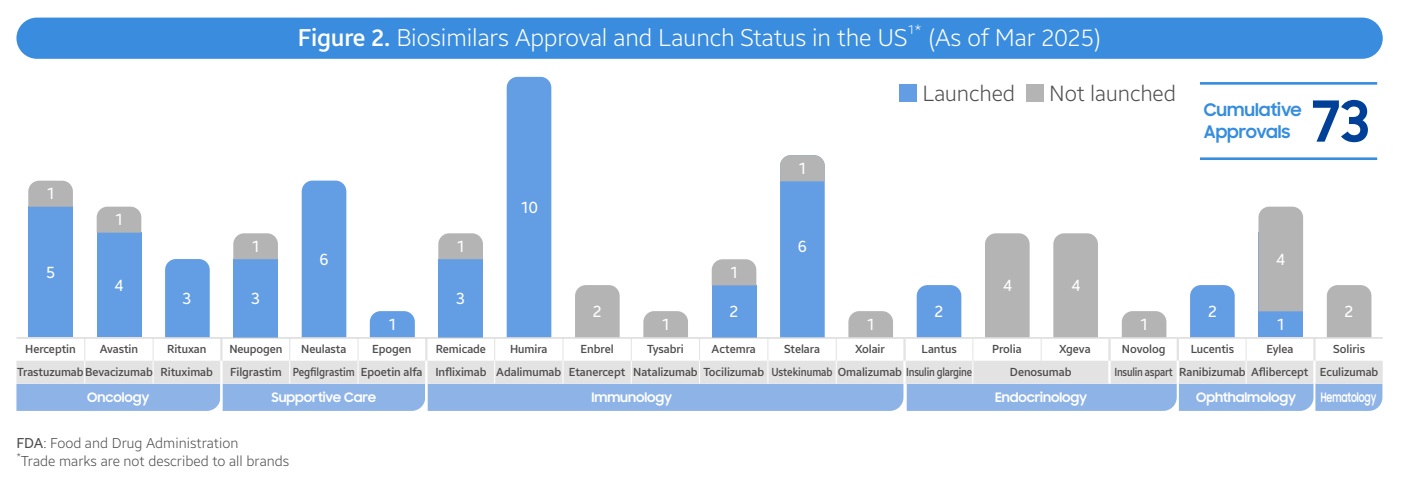
获批后仅有三分之二上市,价格和份额参差不齐
根据报告统计,截至2025年3月,FDA已批准了73个生物类似药,涵盖19个品种。其中,48个生物类似药(占比66%)已在美国市场上市。
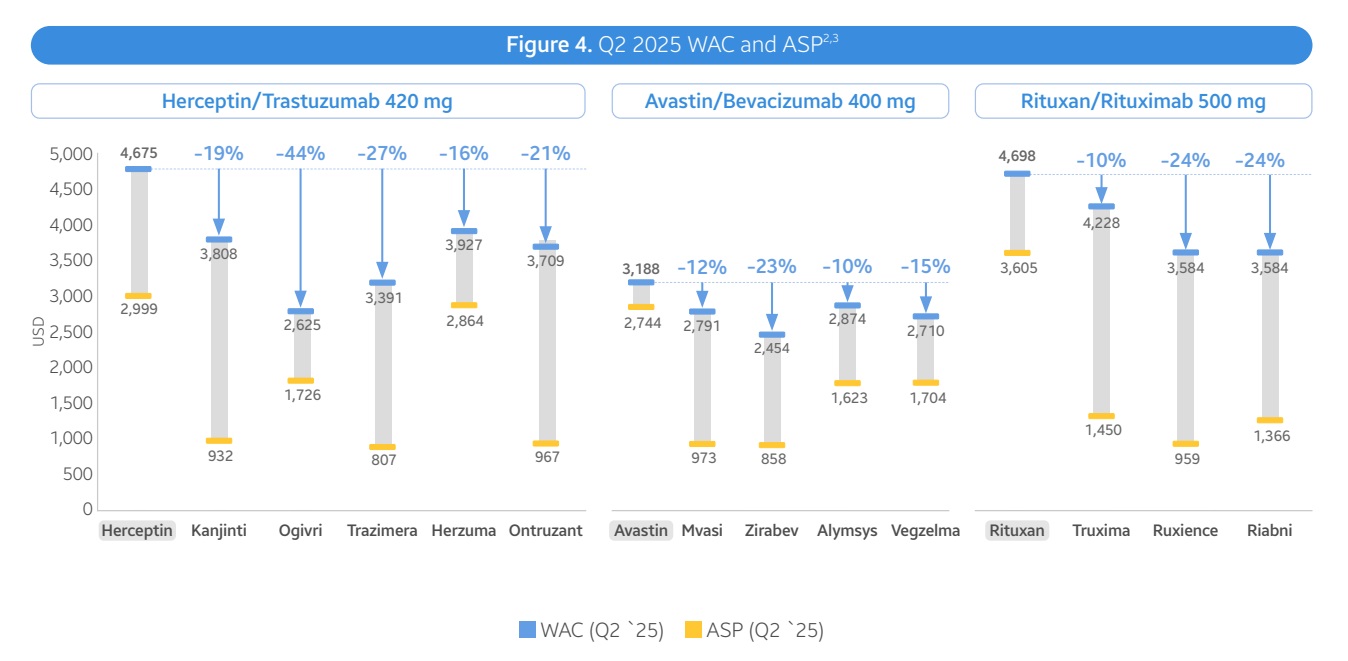
在肿瘤学领域,生物类似药的批发收购成本(WAC)价格较原研药平均折扣10%至44%,平均销售价格(ASP)折扣则达到51%。在支持性治疗领域,生物类似药的WAC价格较原研药折扣17%至67%,ASP折扣平均为35%。
以乌司奴单抗(Ustekinumab)为例,随着Stelara专利到期,多个生物类似药进入市场,其WAC价格较原研药大幅下降超过80%。这不仅为患者提供了更多选择,也为医保系统节省了大量费用。此外,一些生物类似药还通过提供多种WAC选项和无品牌生物药来进一步优化定价策略。
部分生物类似药的推出显著降低了原研药的价格。例如,曲妥珠单抗(Trastuzumab)生物类似药的平均ASP在五年内下降了65%。然而,也有品种出现了价格反弹的现象。例如,由于新上市的生物类似药市场份额较小,其ASP可能反映了WAC定价,导致整体ASP出现上升趋势。
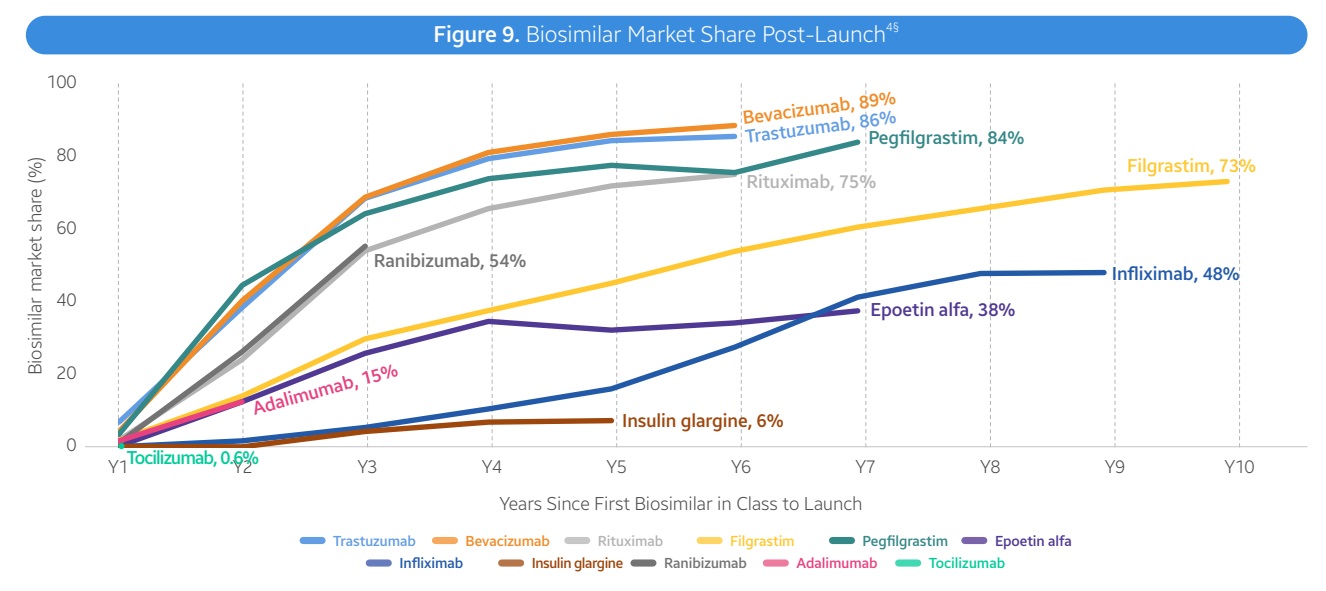
生物类似药的市场份额和价格侵蚀趋势因药物类别而异。例如,在肿瘤学领域,曲妥珠单抗(Trastuzumab)、贝伐珠单抗(Bevacizumab)和利妥昔单抗(Rituximab)的生物类似药市场渗透率较高,五年内平均市场份额达到81%。而在免疫学领域,英夫利西单抗(Infliximab)和阿达木单抗(Adalimumab)的生物类似药市场份额增长较慢,五年内平均市场份额仅为25%。
生物类似药的定价策略也因药物而异。例如,英夫利西单抗的生物类似药在市场上的定价较低,占据了主导地位。而托珠单抗(Tocilizumab)的生物类似药则表现出不同的定价策略,其中Tofidence的ASP仅比原研药低0.3%,而Tyenne的ASP则比原研药低29%。
监管导向是减负、加速、强化竞争
近年来,曾经在生物类似药领域略显保守的FDA致力于简化生物类似药的审批流程,对部分品种提出削减三期临床试验。指南方面,FDA先从可转换研究要求入手,在2024年6月发布了一份更新指南,建议在某些情况下,通过分析、功能和药代动力学(PK)数据即可提供足够的证据,无需进行大规模的转换研究。
与之对应,一贯在生物类似药监管方面较为激进的欧洲药品管理局(EMA)在2025年4月发布了一份反思文件,旨在探索豁免生物类似药进行临床比较有效性研究(CES)的可能性。
报告预期,一旦上述举措均纳入指南,那将显著缩短生物类似药从研发到上市的时间,同时降低了开发成本。例如,过去生物类似药的开发成本通常在1亿至3亿美元之间,开发周期长达7至8年。而随着监管政策的优化,未来生物类似药的开发成本有望降低5000万乃至2.25亿美元,开发周期缩短1至2年。
识林-实木
识林®版权所有,未经许可不得转载
|