首页
>
资讯
>
国际药政每周概要:FDA发布多篇局部药品相同性和生物等效性指南及具体产品指南,临床试验多个终点指南,欧盟药品供应安全结构性对话
出自识林
国际药政每周概要:FDA发布多篇局部药品相同性和生物等效性指南及具体产品指南,临床试验多个终点指南,欧盟药品供应安全结构性对话
2022-10-25
【CMC 与仿制药】
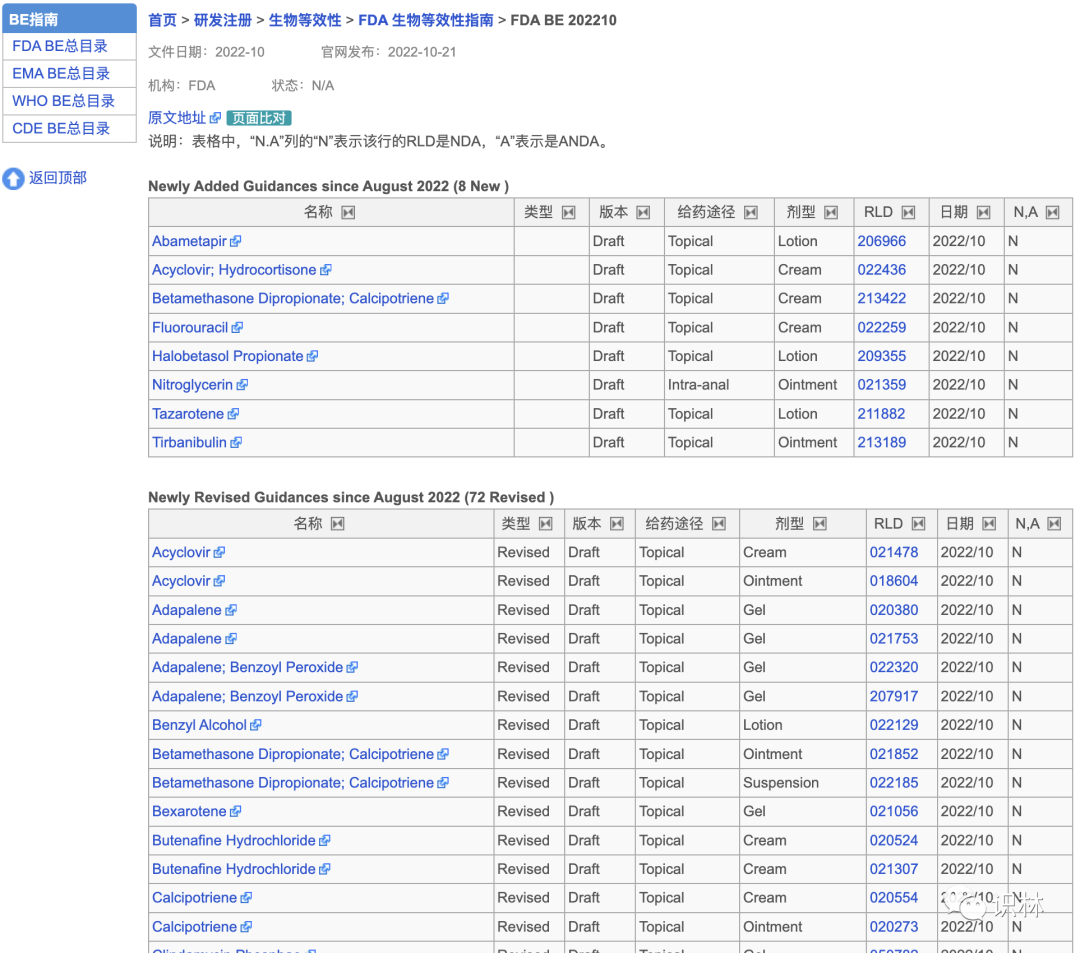
10.21【FDA】BE 指南 新增8篇,修订72篇
FDA 新增和修订的这一批 BE 指南全部为局部软膏、乳膏或凝胶剂。FDA 建议申请人 在考虑设计和开展研究以支持对应用于皮肤的局部作用、液体和/或其他用于皮肤以及外皮和粘膜(例如,阴道粘膜)的半固体产品的 BE 评估时,参考相关产品具体指南以及下面四篇同时发布的局部药品物理化学和结构表征 、体外释放试验研究 、体外渗透试验研究 、体内生物等效性指南 。
登陆识林BE指南数据库 ,阅览来自FDA /EMA /WHO /和CDE 的全部具体产品指南。
10.21【FDA】指南草案 ANDA 提交的局部药品的物理化学和结构(Q3)表征
10.21【FDA】指南草案 ANDA 提交的局部药品的体外释放试验研究
10.21【FDA】指南草案 ANDA 提交的局部药品的体外渗透试验研究
10.21【FDA】指南草案 外用皮肤皮质类固醇:体内生物等效性
10.21【FDA】即将发布的复杂仿制药研发特定产品指南
10.20【FDA】仿制药计划月度绩效活动报告(2022财年)更新
10.17【FDA】ANDA 类的 RS 对应的 RLD 列表
【注册、审评、审批】
10.20【PMDA】药品 PMDA 风险沟通 更新
10.20【PMDA】医疗器械 审评报告 新增 nodoca
10.20【PMDA】药物 修订注意事项 页面更新
10.19【EMA】CTIS 评估时间线 内容更新
10.19【FDA】SOPP 8212: 突破性疗法产品-认定和管理
10.19【FDA】FDA D.I.S.C.O. Burst 版: FDA 批准 Pedmark(硫代硫酸钠)降低1个月及以上儿童局部非转移性实体瘤患者与顺铂相关的耳毒性风险
10.17【FDA】FDA D.I.S.C.O. Burst 版: FDA 批准 Imfinzi(durvalumab)用于局部晚期或转移性胆道癌成人患者
10.14【EMA】寻求科学建议和方案协助申请人指南 内容更新
【创新研发与临床】
10.21【FDA】发布关于临床试验多个终点的最终指南
10.20【FDA】指南定稿 临床研究中多个终点
10.20【FDA】指南摘要播客:临床研究中多个终点
10.20【FDA】临床研究中多个终点 指南快照
FDA 表示,“在药物开发 中进行的大多数临床试验 都包含多个终点,以评估药物疗效并记录药物对一种或多种疾病特征产生有利影响的能力。当在一次试验中分析多个终点时,如果没有对多重性进行适当的调整,药物对一个或多个终点的影响得出错误结论的可能性会增加。”
指南的目的是描述对药物疗效分析的终点进行分组和排序的各种策略,并应用一些公认的统计方法来管理研究中的多重性,以控制对药物疗效做出错误结论的机会。在错误结论的风险没有得到适当控制的情况下根据分析得出结论可能导致关于药物疗效的错误或误导性陈述。详见资讯:FDA就临床试验中多个终点的问题提供指导
10.21【ICH】M11 协同的电子结构化临床试验方案(CeSHarP)
10.21【EMA】临床试验亮点 - 2022年10月
10.21【FDA】指南定稿 神经退行性疾病的人类基因治疗
10.17【FDA】指南定稿 急性髓细胞白血病:开发用于治疗的药品和生物制品
10.17【FDA】指南草案 肿瘤中组织未定性药品的研发
10.17【FDA】指南草案 癌症免疫治疗临床试验中免疫介导的不良反应的特征、收集和报告
10.17【FDA】FDA 局长 Robert M. Califf 在2022年 NORD 突破峰会上的讲话
10.17【FDA】授予19项与罕见疾病(包括ALS)相关的赠款和两份合同
【GxP 与检查】
10.20【FDA】483 比利时 Catalent Belgium SA
10.19【FDA】483 美国 ImClone Systems, LLC
10.19【FDA】483 美国 Compound Preferred LLC
10.19【FDA】483 美国 Hikma Injectables USA Inc
10.18【FDA】警告信 美国 American Cleaning Solutions
10.18【FDA】警告信 中国 北京兴谷绿伞科技有限公司
10.18【FDA】警告信 美国 Forcemech International LLC
10.18【FDA】进口禁令 55-03 更新中国 Chongqing Baijie Changhong Casing Co., Ltd 、四川雄健实业有限公司和郑州元隆化工产品有限公司
10.18【FDA】进口禁令 66-79 新增巴西 Unibeleza Industria E Comercio De Cosmeticos Ltda
10.17【FDA】进口禁令 99-32 新增中国 汕头市橄榄枝生物科技有限公司
10.17【FDA】483 印度 Torrent Pharmaceuticals Limited
【药典相关】
10.21【EDQM】“未来的 CEP”:第二次项目更新
10.21【EDQM】未来的 CEP 项目
10.21【EDQM】CEP 持有人受邀对欧洲药典34.4发表的各论草案进行评议
10.20【EDQM】EDQM 变更关于多晶型化学申请的 CEP 政策
【监管综合】
10.17【EU】全球药品供应链的脆弱性 - 关于药品供应安全的结构性对话
结构性对话的目标是提出一套可能的措施来解决已识别的漏洞,并制定供委员会和欧盟其他当局考虑的政策选择,以确保关键药品、API 和原辅料 的供应保障和可及性。
工作文件的目的是介绍应在供应保障分析中考虑的结构性对话的主要发现,并列出欧盟层面的重点领域,包括新的和正在进行的行动,为提高关键药品、API 和原辅料的供应安全性和可用性的进一步行动提供指导。详见资讯:欧盟发布关于药品供应保障的结构性对话文件,识别供应链脆弱性并确定重点工作领域
10.19【FDA】推进真实世界证据计划
FDA 表示将开始接受申请人 加入新的推进真实世界证据 计划。FDA 长期以来一直在宣扬使用 RWE 来确保产品安全性和有效性的潜力,该计划专门旨在了解如何实现这一目标。FDA 表示,该计划的目的是“提高基于 RWE 的方法的质量和可接受性,以支持新的预期标签 声明,包括已批准产品新适应症 的批准或满足批准后研究要求。”详见资讯:FDA 启动推进真实世界证据计划并更新复杂创新试验设计会议计划
10.21【MHRA】在发现两批 Targocid 200mg被污染后,敦促患者检查包装
10.21【FDA】FDA 综述:2022年10月21日
10.20【FDA】COVID-19 药品和非疫苗生物制品紧急使用授权 页面更新
10.20【FDA】COVID-19 疫苗紧急使用授权 页面更新
10.18【FDA】FDA 综述:2022年10月18日
10.14【EMA】2022年年中报告
【医疗器械】
10.20【FDA】指南草案 选择突破性设备计划指南的更新:减少健康和医疗保健方面的差距
10.17【FDA】CDRH 2023财年拟议指南
识林-Acorn
识林® 版权所有,未经许可不得转载
适用岗位:
注册(RA) :必读。负责理解指南内容,准备和提交科学建议和方案协助请求。研发(R&D) :必读。需要根据指南准备产品开发计划,并在科学建议和方案协助过程中提供专业支持。临床(Clin) :必读。涉及临床试验设计和执行,需遵循指南要求。市场(Mkt) :参考阅读。了解科学建议和方案协助流程,以便在市场策略中考虑监管要求。工作建议:
注册(RA) :根据指南准备科学建议和方案协助请求。 跟踪请求进度,确保及时响应EMA的反馈和要求。 研发(R&D) :根据科学建议调整产品开发计划。 在讨论会议中提供技术支持,解释产品特性和开发策略。 临床(Clin) :设计符合指南要求的临床试验方案。 准备讨论会议,展示临床试验设计和结果。 市场(Mkt) :适用范围:
要点总结:
科学建议和方案协助程序: 明确了获取科学建议或方案协助的流程,指导申请人准备请求,确保请求符合SAWP要求,以便快速有效验证和评估。科学建议和方案协助的范围: 涵盖了质量、非临床和临床方面的问题,包括研究设计、试验和计划,以支持药品的质量、安全性和有效性。费用和减免: 详细说明了科学建议的费用结构和支付时间,以及孤儿药和中小企业的费减免政策。申请和验证流程: 描述了通过IRIS平台提交申请、准备简报包的结构和内容,以及请求的验证流程。讨论会议和后续行动: 指导申请人如何准备讨论会议,以及科学建议的后续行动,包括澄清和跟进请求。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
临床(Clinical) :必读。应根据文件指导设计临床试验方案,确保试验设计的科学性和合规性,特别是在选择终点指标和评估疗效时。研发(R&D) :必读。在新药开发过程中,需参考文件中关于剂量选择、治疗计划和药物相互作用的指导,以优化药物设计和剂量方案。注册(Regulatory Affairs) :必读。负责确保提交给FDA的所有文件和数据符合指南要求,包括IND申请和市场申请。质量保证(QA) :必读。需监督临床试验的执行,确保符合指南中的质量管理要求。适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证):负责确保IVPT方法的合规性和质量控制。 研发(R&D):负责IVPT方法的开发、优化和验证。 注册(Reg):负责ANDA提交,需理解IVPT要求以支持申报文件。 临床(Clin):虽然不直接参与IVPT,但需了解IVPT结果以评估临床试验设计。 工作建议:
QA:监控IVPT方法的实施,确保符合FDA指南要求,进行必要的质量审核。 研发:系统地开发和优化IVPT方法,进行方法验证,确保方法的适用性和灵敏性。 注册:在ANDA中整合IVPT数据,确保申报材料符合FDA要求。 临床:参考IVPT结果,设计临床试验以验证药物的生物等效性。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证) :应深入理解IVRT方法验证和样品分析方法验证的要求,确保实验过程符合FDA指南和USP<1724>的要求。研发(R&D) :在开发IVRT方法时,应依据本指南选择合适的膜材料、受体溶液,并优化实验参数。注册(Regulatory Affairs) :需熟悉本指南,以便在提交ANDA时,能够提供满足FDA要求的IVRT研究资料。临床(Clinical) :在生物等效性研究中,需要参考IVRT研究结果来设计临床试验。工作建议:
QA :监控IVRT方法开发和验证过程,确保所有步骤符合质量管理体系要求,包括设备校准、样品处理和数据记录。R&D :在方法开发阶段,应详细记录不同膜材料和受体溶液的比较数据,以及对实验参数优化的尝试和结果。注册 :在准备ANDA文件时,确保包含所有必要的IVRT研究资料,并按照eCTD格式要求提交。临床 :利用IVRT研究结果来预测药物释放特性,为临床试验设计提供科学依据。适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
临床研究(Clinical):必读。在设计和执行临床试验时,需考虑多个终点的统计方法和策略,以控制I型错误率。 统计学(Statistics):必读。负责选择合适的统计方法来处理多个终点问题,并为临床团队提供统计支持。 注册(Registration):必读。在准备药品注册文件时,需理解多个终点的监管要求,确保临床试验结果符合法规标准。 工作建议:
临床研究:在设计临床试验方案时,明确主要终点和次要终点,并与统计团队合作,选择合适的多重比较校正方法。 统计学:为临床团队提供统计咨询,帮助他们理解不同多重比较校正方法的适用性和影响,确保试验结果的可靠性。 注册:在撰写注册文件时,详细描述临床试验中使用的多重比较校正方法,并解释其对试验结果的影响。 适用范围:
要点总结:
多重性问题 :强调在临床试验中,当分析多个终点时,需要适当调整以控制I型错误率,避免错误结论。终点层级 :明确了主要终点、次要终点和探索性终点的层级结构,以及它们在支持药品效果结论中的作用。共主终点 :讨论了在某些疾病中,需要在两个或更多不同的终点上证明治疗效果,以建立临床效益。复合终点 :描述了将多个临床结果合并为单一变量的情况,以及分析和报告这些复合终点时的考虑因素。多重比较校正方法 :提供了多种统计方法,用于控制多重性问题,包括Bonferroni方法、Holm程序和Hochberg程序等。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
临床(Clinical) :必读。在设计临床试验时,应考虑如何合理选择和报告多个终点,确保试验结果的科学性和合规性。研发(R&D) :必读。在药物开发过程中,需依据指南评估多个终点的合理性,优化临床试验设计。注册(Regulatory Affairs) :必读。在提交临床试验申请时,需确保终点选择和报告符合FDA指南要求。适用范围:
文件要点总结:
终点选择与报告: 明确指出在临床试验中选择和报告多个终点时应遵循的原则和标准。统计考量: 强调了在多个终点分析中统计学考量的重要性,包括对多重比较的控制。监管沟通: 鼓励在临床试验设计阶段与FDA进行沟通,以确保终点选择和报告的合规性。数据完整性: 规定了在报告多个终点时,如何保证数据的完整性和透明度。适应性设计: 讨论了适应性设计在多个终点分析中的应用,以及其对试验结果的影响。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证) 注册(药品注册) 研发(药品研发) 临床(临床研究) 工作建议:
QA:确保所有Q3表征测试符合FDA指南要求,并监控测试结果以保证产品质量。 注册:在提交ANDA时,包含必要的Q3表征数据以证明仿制药与参比制剂的等效性。 研发:进行Q3表征测试,以全面了解产品特性,并支持BE研究。 临床:在设计临床试验时,考虑Q3表征结果对生物等效性的影响。 适用范围:
要点总结:
Q3表征目的: 明确了Q3表征用于识别仿制药的剂型和描述可能影响产品性能的关键属性。Q3表征类型: 区分了基本Q3表征(描述剂型)和全面Q3表征(详细描述关键属性)。Q3比较: 强调了比较仿制药与参比制剂Q3属性的重要性,以支持生物等效性(BE)的证明。Q3属性的比较结果: 定义了Q3相同性、Q3相似性和Q3差异性的概念,并讨论了它们对BE证明的影响。与FDA的沟通: 鼓励ANDA申请者在开发过程中与FDA沟通,以确保Q3表征方法和BE策略的适宜性。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
“注册”(RA):必读。负责ANDA提交,需理解指南要求,确保申报资料符合FDA规定。 “临床”(Clin):必读。负责设计和执行体内生物等效性研究,需遵循指南中的研究设计和数据分析要求。 “QA”:必读。负责确保研究和生产过程符合FDA规定,需监督指南的实施情况。 工作建议:
“注册”(RA):在准备ANDA时,应详细审查本指南,确保所有要求得到满足,并在申报资料中体现。 “临床”(Clin):在设计和执行研究时,应严格遵循本指南中的研究设计、受试者筛选、数据收集和分析等要求。 “QA”:在监督研究和生产过程中,应确保所有操作符合本指南的规定,及时发现并纠正偏差。 适用范围:
要点总结:
适用岗位及工作建议:
临床(Clinical) :必读。应熟悉ICH M11指南中关于临床试验方案的结构和内容要求,确保临床试验方案的制定和执行符合国际标准。注册(RA) :必读。需掌握ICH M11指南中电子结构化临床试验方案的技术规范,以便在药品注册过程中与监管机构有效沟通。研发(R&D) :必读。应了解ICH M11指南中关于临床试验方案设计的一般原则,以指导新药研发过程中的临床试验方案设计。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。