|
首页
>
资讯
>
印度Lupin Limited各工厂近年警告信分析
出自识林
印度Lupin Limited各工厂近年警告信分析
2022-12-05
近年来印度Lupin公司成为了FDA警告信的常客,在三家印度工厂2017年和2019年收到警告信后,2021年,美国新泽西州的Novel Laboratories也收到了警告信。随后在今年9月,又一家印度原料药工厂(Lupin Limited MIDC)收到警告信。下表总结了Lupin各工厂近年来收到的警告信和其中所涉及的缺陷项。
| 年份
| 工厂
| 地区
| 产品 类型
| 缺陷项
|
| 2022
| Lupin Limited MIDC
| 印度
| 原料药
| 1. 未能制订并遵守关于设备清洁及允许其用于中间体和API生产的合适的书面规程。
2. 未能制订书面规程来监测和控制那些可能导致中间体和API药质量属性变异的工艺的进程和表现。
3. 未能调查所有严重违规行为。
|
| 2021
| Novel Laboratories, Lupin
| 美国
| 制剂
| 1. 未能制订并遵守充分的设备清洁和维护的书面规程。
2. 未能制订充分的书面生产和工艺控制规程,以保证药品所声称的或表明拥有的成分、规格、质量和纯度。
3. 未能为质量部门制订并遵守足够的书面职责和规程。
|
| 2019
| Lupin Limited Unit I
| 印度
| 原料药/制剂
| 1. 未能彻底调查任何未解释的差异或任一批次的失败或原辅料不符合其任一质量标准,不管该批次是否已经放行销售。
2. 未能制订充分的书面生产和工艺控制规程,以保证药品所声称的或表明拥有的成分、规格、质量和纯度。
3. 未能做到间隔适当时间,清洁、维护设备和器具,根据药品特性进行消毒和/或灭菌,防止出故障与污染,以免改变药品的安全性、成分、规格、质量或纯度而超出官方或其它既定要求。
|
| 2017
| Lupin Goa
| 印度
| 原料药
| 1. 未能彻底调查任何未解释的差异或任一批次的失败或原辅料不符合其任一质量标准,不管该批次是否已经放行销售。
2. 未能为生产每一步操作设定适当的时限,以保证药品质量。
|
| 2017
| Lupin Indore
| 印度
| 制剂
| 1. 未能为生产每一步操作设定适当的时限,以保证药品质量。
2. 未能彻底调查任何未解释的差异或任一批次的失败或原辅料不符合其任一质量标准,不管该批次是否已经放行销售。
|
下表将这几封警告信中重复出现的缺陷项进行了汇总。通过对比可以发现,FDA对于同一公司下的工厂,经常会发布类似的缺陷项,这一方面是由于同一集团下的工厂质量体系存在类似的问题,另一方面是由于FDA在检查前的准备工作中,往往会事先回顾该公司下的工厂以往检查中所涉及的缺陷项,这些缺陷项将是下一次检查的重点。这一理念也是FDA所一直强调的“使用基于风险的模型来决定接下来要检查哪些公司”。
| 211条款
| 缺陷项
| 存在此缺陷项的工厂
|
| 21 CFR 211.192
| 未能彻底调查任何未解释的差异或任一批次的失败或原辅料不符合其任一质量标准,不管该批次是否已经放行销售。
| Lupin Goa(2017)、Lupin Indore(2017)、Lupin Limited Unit I(2019)、Lupin Limited MIDC(2022)
|
| 21 CFR 211.111
| 未能为生产每一步操作设定适当的时限,以保证药品质量。
| Lupin Goa(2017)、Lupin Indore(2017)
|
| 21 CFR 211.100(a)
| 未能制订充分的书面生产和工艺控制规程,以保证药品所声称的或表明拥有的成分、规格、质量和纯度。
| Lupin Limited Unit I(2019)、Novel Laboratories, Lupin(2021)、Lupin Limited MIDC(2022)
|
| 21 CFR 211.67(a)
| 未能做到间隔适当时间,清洁、维护设备和器具,根据药品特性进行消毒和/或灭菌,防止出故障与污染,以免改变药品的安全性、成分、规格、质量或纯度而超出官方或其它既定要求。
| Lupin Limited Unit I(2019)、Novel Laboratories, Lupin(2021)、Lupin Limited MIDC(2022)
|
| 21 CFR 211.22(d)
| 未能为质量部门制订并遵守足够的书面职责和规程。
| Novel Laboratories, Lupin(2021)
|
重点缺陷项问题分析
细读各缺陷项下的细节描述,可以发现,Lupin各工厂的主要问题集中在两个方面:无效OOS和设备清洁验证,并且相似的缺陷项在各工厂的检查中反复出现。下文分析了无效OOS的主要问题和原因,对于清洁验证等更多细节问题的分析,可参见识林案例解析“印度Lupin Limited各工厂近年警告信分析”。
OOS调查和无效OOS
4家接受检查的工厂都出现了对超标结果(OOS)分析调查不充分,在没有确认根本原因的情况下关闭OOS调查,并以重新测试结果放行或拒绝批次,也未能实施有效的纠正和预防措施(CAPA)。
下表为FDA在2017年Lupin Goa的483中统计的无效OOS比例。商业化成品,一年中共有89次OOS调查,其中,通过调查将OOS无效的有67个,OOS无效率为75%;稳定性研究中共有31次OOS,经调查无效化的有30个,无效率高达97%;而对于原料,共有48个OOS,经调查无效化的有34个,无效率为71%。这意味着该企业避免向 FDA 发送现场警报:如果确认 OOS 结果,可能会触发批次召回。
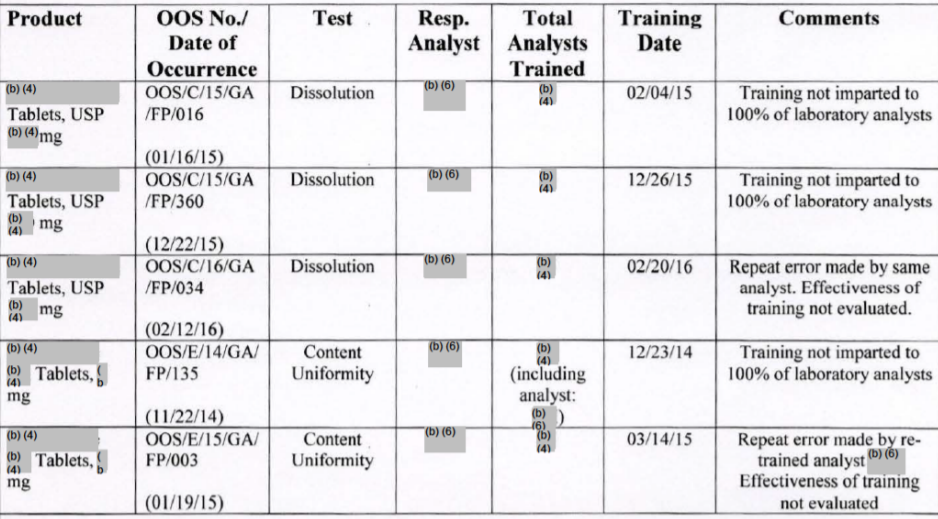
极高的OOS无效化比例,有两种可能性,一是真OOS,但不想体现出来,所以,让人为错误、实验室错误来背锅。二是确实检验问题很大,人为错误层出不穷。因此,在解释了上述将OOS无效化的调查中存在的不足和问题以后,FDA在483中又汇总了一个关于OOS的培训汇总表(见下表)。
从表中可以看出:这里有3个是片剂溶出度不合格的OOS,另外两个是片剂含量均匀度的OOS。经无效化后,认定是检验人员的错误,于是,相应的整改措施,包括对人员进行培训。
汇总表里列出了该参加培训的人员数量,和实际被培训的人员数量。虽然具体数量未公开,但是在备注里汇总的注释包括以下3种情况:
- 经过再培训的检验员,又出现重复的错误,导致假OOS发生。
而在上文中也提到,Lupin另外两家工厂在2019年和2022年的警告信中重复出现了此问题,说明此类问题一直没有得到有效的整改。多场地重复违规说明公司对药品生产的监管和控制不足,这些重复出现的问题也将是FDA今后对此公司下属工厂检查的关注点。印度Lupin的警告信对我国的制药企业也是一个警示,企业必须立足日常生产,对质量管理体系做主动、持续的改进,将偏差调查和CAPA落实到位,避免反复出现同样的问题。
作者:识林-雪杉
识林®版权所有,未经许可不得转载
必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。
- 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。
- 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。
适用范围:
本文适用于涉及化学药、生物制品、疫苗和中药等药品类型的企业,包括创新药、仿制药、生物类似药和原料药等注册分类。适用于不同规模的企业,如Biotech、大型药企、跨国药企、CRO和CDMO等,由相关药品监管机构发布。 文件要点总结: - 质量管理体系概述:明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。
- 产品质量实现要素:涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。
- 质量保证要素:包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。
- 质量风险管理:介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。
- 质量管理系统文件:规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |