|
首页
>
资讯
>
FDA在2022财年发布的警告信和483缺陷项统计梳理
出自识林
FDA在2022财年发布的警告信和483缺陷项统计梳理
2022-11-25
本文对FDA在2022财年(2021年10月1日至2022年9月30日)发布的警告信和483中所涉及的所有缺陷项按照类别和出现频率进行了统计梳理,从而帮助业界更清楚地了解FDA检查的最新关注点和GMP检查中最常出现的缺陷项,进而有针对性地进行自检和改进工作。
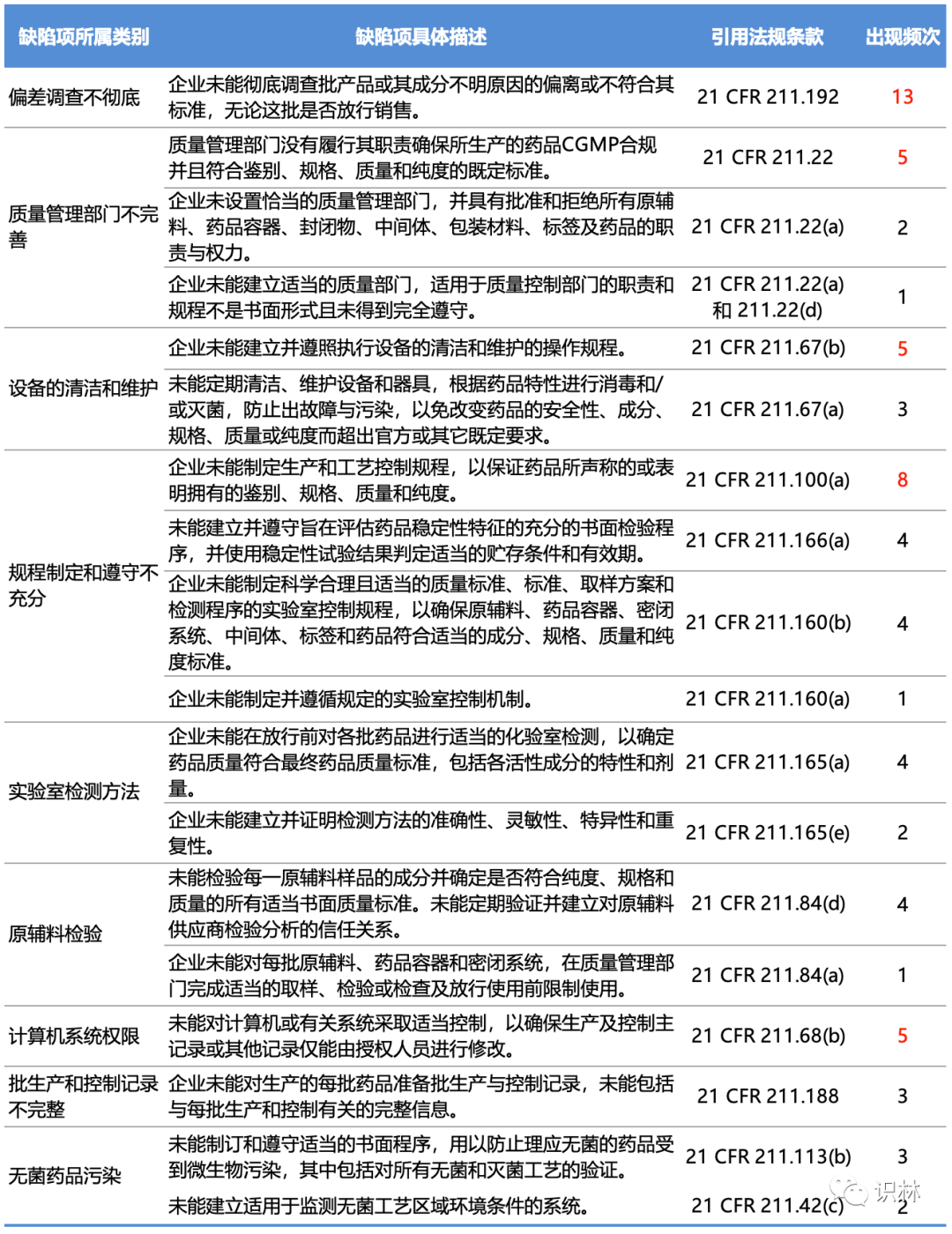
警告信中的缺陷项
以识林当前已经翻译的32封FDA在2022财年发布的药品相关警告信为样本,其中包括制剂25封,原料药6封,生物制品1封。这32封警告信中共涉及92条缺陷项,下表将出现频次较高的缺陷项划分了几个主要类别,并统计了各缺陷项所对应的21 CFR 211条款和出现频次(标注红色的为出现频次较高的缺陷项)。
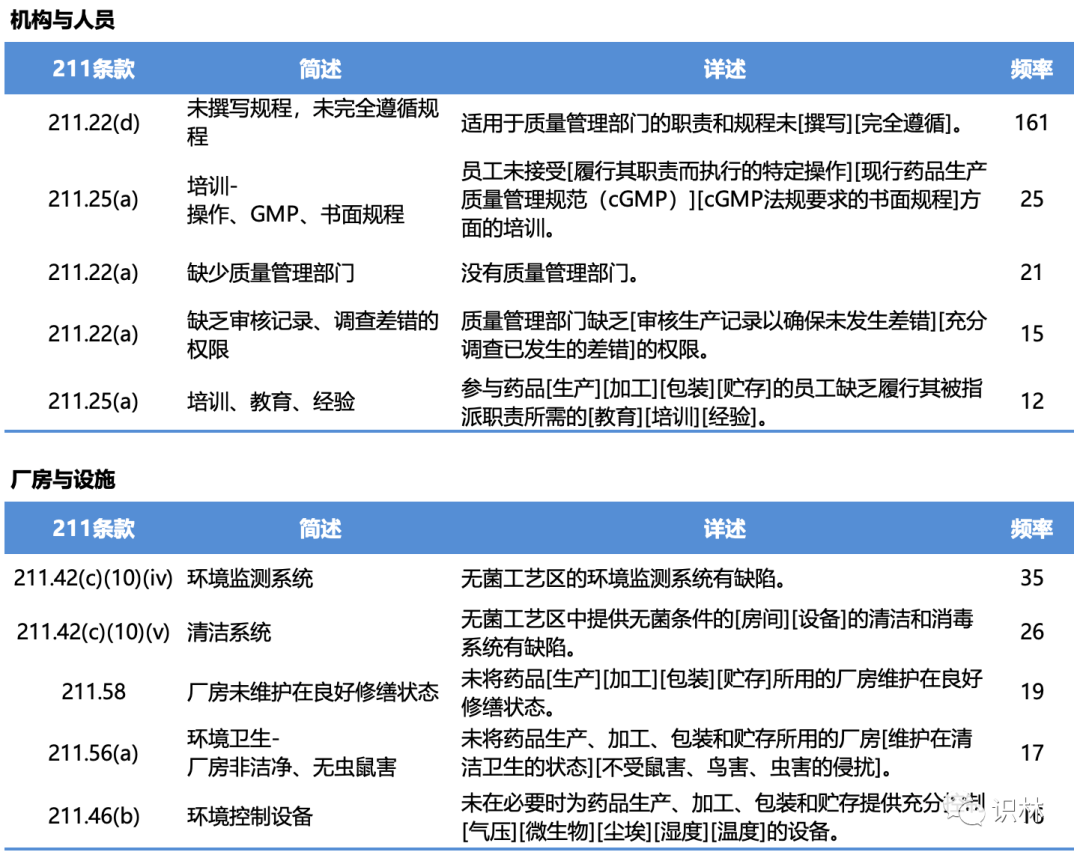
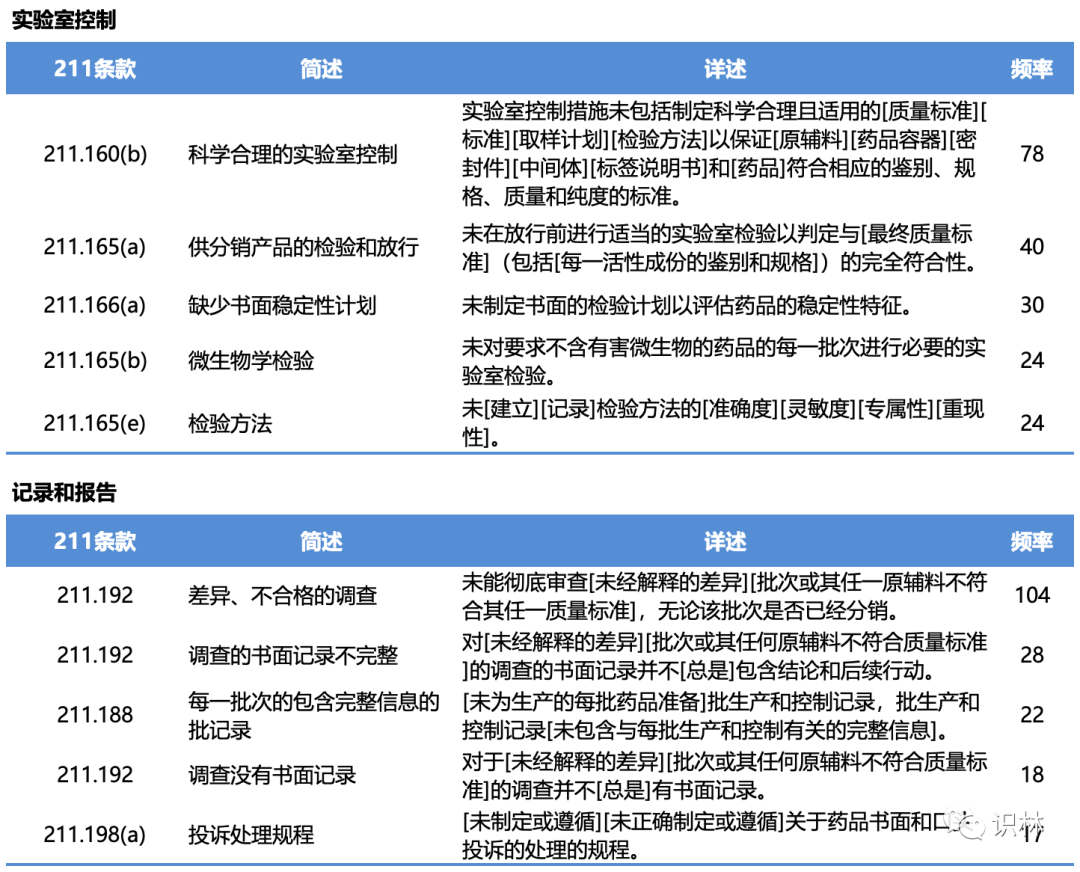
483中的缺陷项
在2022年财年,FDA与药品有关的现场检查,有483缺陷的总计466次;与生物制品有关的现场检查,有483缺陷的总计61次;与医疗器械有关的现场检查,有483缺陷的总计528次。其中与211有关的缺陷项分类、简述、详述、频率以及具体条款,见下列各表。(注:下列各表中仅列举各类别中出现频率较高的缺陷项类别,并非所有483缺陷项)
识林-雪杉
识林®版权所有,未经许可不得转载
适用岗位必读指南: - QA:负责确保所有操作符合cGMP要求,包括生产、质量控制、设备维护等。
- 生产:必须遵守书面程序,确保产品质量。
- 质量控制(QC):负责样品的测试和批准或拒绝,以及稳定性测试。
- 设备维护:确保设备清洁、维护和校准符合规定。
- 仓储与分销:遵守药品存储和分发的书面程序。
文件适用范围:
本文适用于美国市场的所有成品药品的cGMP(现行良好生产规范),不包括正电子发射断层扫描药物。适用于化学药品、生物制品、疫苗、中药等药品类型,包括创新药、仿制药、生物类似药、原料药等注册分类。适用于所有在美国运营的Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 文件要点总结: 质量控制单元的责任: 必须有一个质量控制单元,负责批准或拒绝所有组件、药品容器、包装材料、标签和药品,并审查生产记录以确保没有错误发生或错误已得到全面调查。 人员资质与责任: 参与药品生产、加工、包装或储存的人员必须具备相应的教育、培训和经验,并遵守良好的卫生习惯。 设备设计、清洁与维护: 设备应适当设计,便于操作、清洁和维护,并按规定进行定期清洁和维护。 组件和药品容器的控制: 必须有书面程序详细描述组件、药品容器和闭合件的接收、识别、存储、取样、测试和批准或拒绝。 生产和过程控制: 必须有书面程序确保药品具有其声称或代表的身份、强度、质量和纯度,包括偏差处理和产量计算。 包装与标签控制: 必须有书面程序确保正确的标签和包装材料用于药品,包括防篡改包装要求。 仓储与分销程序: 必须有书面程序描述药品的存储和分发,确保药品质量。 实验室控制: 必须建立科学合理的规格、标准、抽样计划和测试程序,以确保药品及其组件符合适当的身份、强度、质量和纯度标准。 记录与报告: 所有与生产、控制或分发相关的记录必须保存至少一年,或在特定情况下保存更长时间,并随时可供授权检查。 退回和报废药品的处理: 退回的药品必须被识别并保留,除非证明其符合适当的安全、身份、强度、质量和纯度标准,否则应销毁。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |