首页
>
资讯
>
全球主要药监机构 AI 监管的进展与方法概览
出自识林
全球主要药监机构 AI 监管的进展与方法概览
2025-08-08
以Deepseek和ChatGPT为代表的大语言模型(LLM),推动了人工智能(AI) 技术在制药行业的应用热潮。AI技术在药物发现、非临床研究、临床试验以及上市后监管等多个环节展现出巨大潜力,同时也为全球药监机构带来了前所未有的挑战。
如何让能力卓越的通用型LLM(如Deepseek)掌握制药行业的专业知识,并能够按药监或药企日常的工作场景赋能,是AI在制药专业领域垂直应用的核心问题。
识林不仅在探索AI与结构化知识库结合辅助药品研发 注册 、生产质量与监管的落地场景,也在持续关注全球主要药监机构发布的制药AI相关文件和动态。近期在国家、省市监管机构和企业、学协会等多次分享交流 ,在交流过程中深切感受到国家和各省药监部门已经在积极拥抱AI。对于药企,在努力探索药品生命周期 的各个阶段充分利用AI的同时,也应对其监管和合规有充分的考量和交流。识林将上述内容整理形成本文,期待给读者一些参考。
全球制药AI监管图景:法案、指南、规划与实践
AI监管的历史远长于LLM的兴起,出于聚焦和精要的考量,本文的讨论范围限定于2022年11月ChatGPT发布之后,全球主要监管机构(中国NMPA,美国FDA,欧盟EMA,以及ICH 、WHO和PIC/S)的官方发布的文件,且文件主题必须与AI直接相关。考虑到实际情况,本文也会涉及普适性的官方文件,如欧盟的AI法案等,这些文件虽然不专门针对药品监管,但在法律和法规层面引导制药AI监管体系的建立。
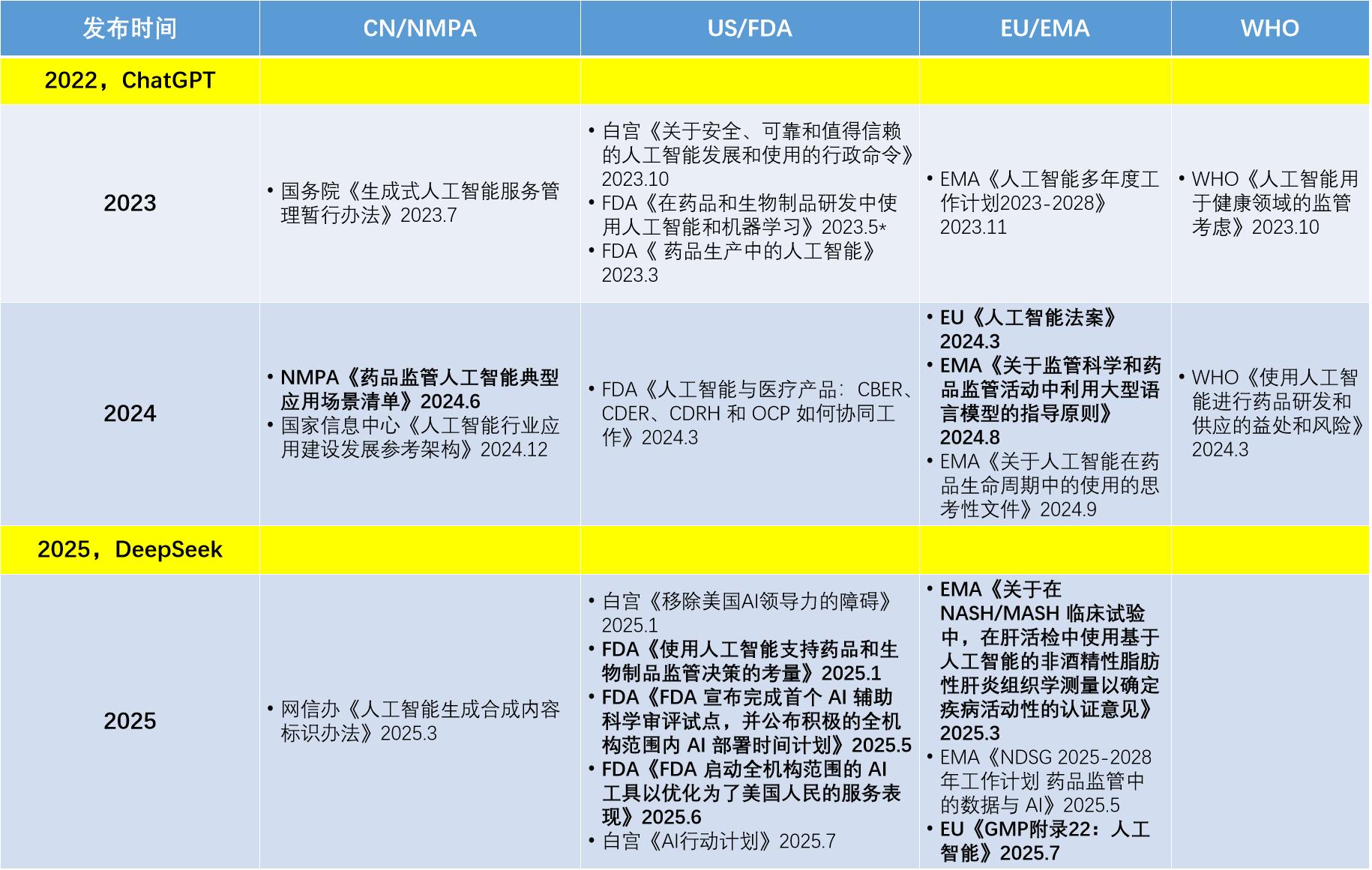
从文件发布时间角度(图1)可以看到,尽管AI并不限于LLM,但自从ChatGPT面世以来,各监管机构很明显加快了AI监管文件的发布节奏,其中有许多文件针对的正是LLM。ICH暂未发布与AI直接相关的指南。而PIC/S仅随同EU更新了GMP附录22人工智能 ,因此未在表中列出。
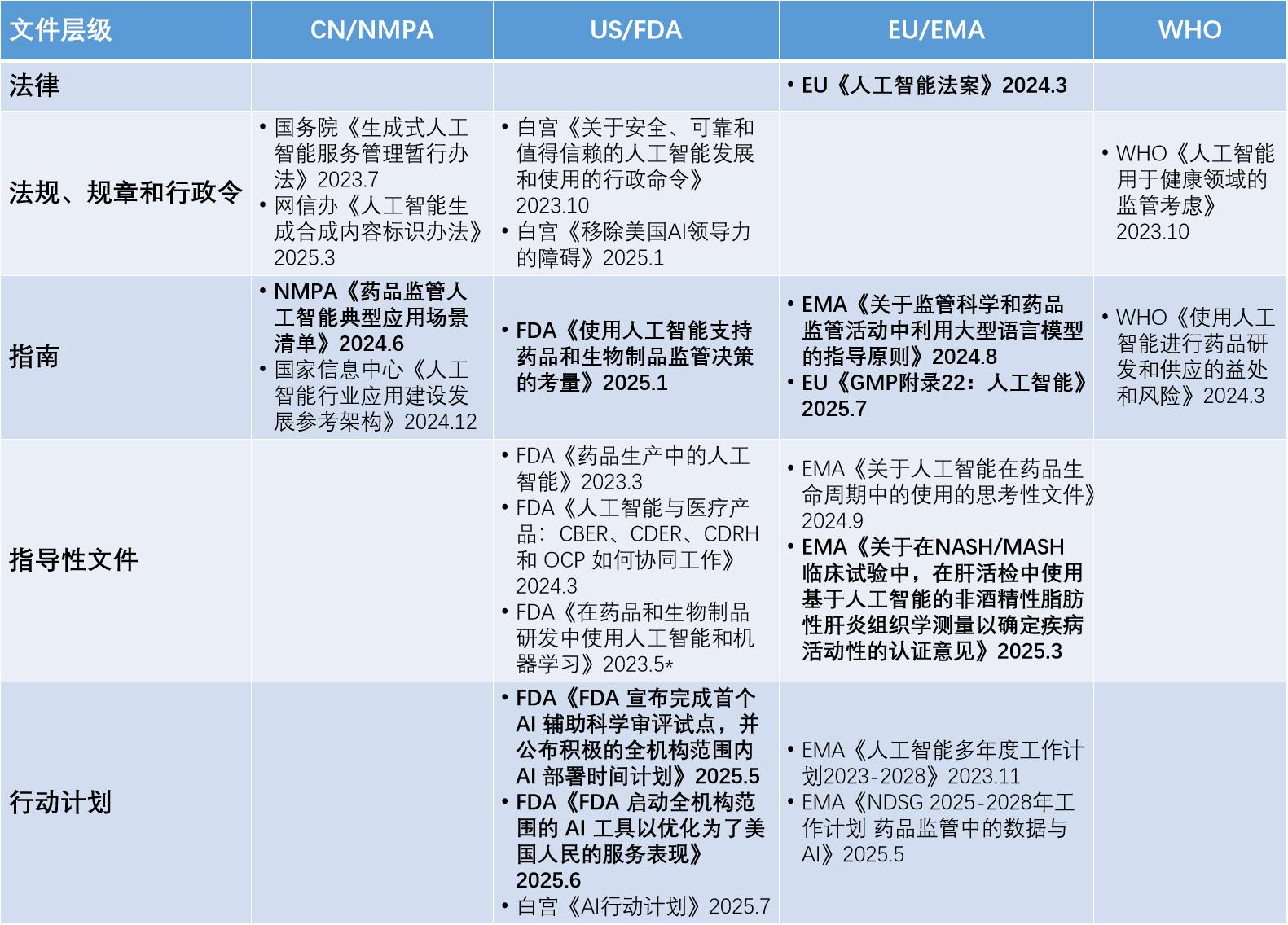
从文件层级角度(图2),不同地区和国际组织发布的有关AI应用的官方文件各有侧重。在立法层面,欧盟走在前列。2024年3月,欧盟率先发布第一个针对AI的法律文件——《人工智能法案》,为AI技术应用提供了全面的法律框架。
国家政策层面,我国和美国均有政策文件指导AI技术发展。我国由国务院和主要部委规划AI技术发展方向,已于2023年7月发布针对性规范性文件《生成式人工智能服务管理暂行办法》 ,并在2025年3月发文提出AI生成内容标识。美国通过行政命令指导AI技术发展方向,但两届政府的行政令之间却针锋相对,相比拜登,特朗普倾向于更为激进的“去监管”。在2025年7月,白宫基于特朗普的行政令发布了一份《AI行动计划》 ,一口气提出了90多项新的联邦政策倡议,其中提及FDA应设立监管沙盒(regulatory sandbox)以促进AI应用。
指南层面,欧盟EMA和美国FDA分别发布了以AI为标题的正式指南文件。欧盟更是在7月7日推出了全球主要监管市场的第一份官方AI GMP指南 。尽管我国NMPA和ICH也发布了如“模型引导的药物开发” 、“机器学习(ML)”等涉及AI技术的相关指南文件,但通常不以AI为主题。
指导性文件层面,我国NMPA、美国FDA、欧盟EMA、WHO均有与AI相关的文件出台。ICH虽然发布了M15(模型引导的药物研发) 等与AI有关的指南,但尚未发布以AI为标题的文件。
主要药监机构的AI定义:自主,学习,推理
欧盟AI法案中特别强调,AI涵盖多个学科和大量场景,术语被广泛使用,因此其定义需要予以特别强调。这不仅是确保监管的确定性、促进国际融合协调和广泛接受的关键,同时也是适应AI领域快速技术发展的必要条件。AI的定义应与全球主要监管机构在AI 方面的工作紧密对齐,基于区分AI系统与传统软件系统或编程方法的关键特征,并排除那些仅基于人类定义规则、自动执行操作的系统。
欧盟、美国FDA和WHO都提供了各自对AI的定义(图3),这些定义在某些方面有共通之处,但也存在差异。截至目前,我国药品监管体系的官方文件中尚未见AI的定义。
欧盟AI法案是目前全球各级监管主体发布的关于AI的最高级别法律文件,其对“AI系统”的定义强调了AI系统的自主性和适应性,以及其从输入中推断如何生成影响环境的输出的能力,涵盖了AI的广泛应用。
FDA的AI定义更侧重于AI系统的目标导向性,以及其在感知、模型抽象和推理方面的能力。此外,FDA的定义特别提到了ML作为AI的一个重要子集,并强调了数据在AI性能提升中的作用。
WHO的定义将AI定位为计算机科学、统计学和工程学的交叉领域,强调了算法或模型在执行任务和展示行为方面的能力。WHO的定义突出了AI的学习能力,尤其是在没有明确编程的情况下通过数据进行学习。此外,与FDA定义类似,WHO也明确提及了ML。
对比这三个定义可以看到,尽管它们都强调了AI的自主性和学习能力,但各有侧重点。欧盟的定义较为抽象但更加全面;FDA的定义更具体,且侧重应用;而WHO的定义则更强调AI的跨学科特性。这些差异除了反映了不同监管机构对AI应用的不同关注点和监管重点,也能帮助其他监管部门分辨如今涵义愈加宽泛的AI一词,从而针对AI与非AI进行合理的监管。
制药AI监管:两个维度和两条路径
在探讨全球药品监管机构的AI监管实践中,可从两个维度入手。“将AI用于监管”指的是药监机构作为AI的使用者,将AI技术视为一种监管工具,将其融入药品审评、检查等监管流程之中,以提升监管效率。而“对AI进行监管”指的是药监机构作为AI的监管者,对药品生命周期 内AI技术的运用进行有效监管,旨在确保在AI技术不断演进的同时,对药品的安全性、有效性和质量控制 可能带来的风险进行严格管控。
将AI用于监管:中国场景引导,EMA谨慎推进,FDA的Elsa
中国和欧盟药监机构专门发文指导AI在药品监管中的应用,而FDA则高调地推出基于LLM的AI工具Elsa。
NMPA发布的《药品监管人工智能典型应用场景清单》 明确了15个具有示范性的应用场景,覆盖了从药品准入审批到日常监管,再到服务公众和辅助决策的全方位监管流程。
NMPA的设想是,在准入审批类应用中,AI可被用于形式审查和辅助审评,通过构建LLM,实现对药品和医疗器械 注册申报材料的自动化智能审查,提高审查效率和质量;在日常监管类应用中,AI可构建生产检验数据靶向分析模型和远程监管风险预警模型,为监管部门提供了强有力的数据支持,此外还可辅助现场检查的准备工作,并在检查后辅助撰写检查报告,提高报告撰写的效率和质量;服务公众类应用场景中,AI可通过嵌入业务数据和政策法规的LLM,提升面向公众的业务办理、信息查询、政策咨询服务质量;在辅助决策类应用中,AI可进行业务数据查询、数据分析 与预测,以及工作方案研究,为监管决策提供有力支持。特别是在风险管理 领域,AI技术通过对药品品种档案、药品安全信用档案等业务数据进行信息检索和智能分析,可实现对药品监管全生命周期 中潜在风险的精准识别和预警等等。
EMA的《关于监管科学和药品监管活动中利用大语言模型的指导原则》 为欧盟药品监管机构中的用户提供了一个关于如何安全、负责任和有效地使用LLM的框架。不同于NMPA从应用场景出发,欧盟文件聚焦于LLM本身的特性和能力,涵盖了LLM的定义、分类,强调了LLM在自动化信息处理、知识挖掘、虚拟助手和聊天机器人中的关键作用,并特别指出了LLM在药品监管中的潜在用途,如撰写辅助、信息搜索与总结、语言翻译、教育辅导和编程语言支持。
但EMA更有参考价值的AI文件不是指南,而是其在3月发布的首个基于AI的创新药物开发方法的资格认定意见 。这一AI工具名为AIM-NASH,它能够帮助病理学家分析肝脏活检扫描图像,以确定代谢功能障碍相关脂肪性肝炎(MASH,此前称为非酒精性脂肪性肝炎,NASH)的严重程度。EMA在经过公开征求意见后,发布了对AIM-NASH工具的资格认定意见,这意味着药监部门明确表示可以接受该工具生成的证据作为未来申请中的科学证据。目前该工具也在FDA的“新药创新科学和技术方法” (ISTAND)的待认证列表中。
FDA应用AI的进程在新局长Makary上任之后明显加快。5月8日,其宣布将在6月全面推广AI工具 ,以协助进行科学审评工作。Makary指示所有FDA中心立即开始部署AI辅助审评系统,目标是缩短科学审评过程中任务的执行时间。FDA表示将推行一个“激进的时间表”,以在6月30日之前实现AI工具在各中心的“全面整合”。不到一个月,6月2日,FDA就正式发布生成式AI工具Elsa ,旨在助力从科学审评人员到检查人员的工作效率提升。据FDA新闻稿称,Elsa的核心功能包括阅读、写作和总结。在实际应用中,它能够对不良事件 进行总结,以支持安全性评估;快速进行标签比较;生成代码,帮助开发用于非临床应用的数据库等。这些功能只是Elsa在提升药监运营效率方面应用的一小部分示例。
FDA对Elsa的细节透露甚少,但FDA人员近期发布了一篇文献《利用生成式人工智能进行FDA指南文件的语义搜索》 ,该研究的核心目标与Elsa的功能高度契合,正是评估生成式AI结合检索增强生成(Retrieval-Augmented Generation,RAG)工具是否能够准确回答审评人员有关FDA指南文件内容的问题,其方法和结论颇具参考价值。最终结果显示,从112份精心挑选的临床审评所需指南文件中,结合RAG的GPT-4 Turbo在33.9%的情形下生成正确回答并提供额外的有用信息;35.7%生成正确回答;17.0%生成的回答包含部分正确信息;而13.4%生成的回答包含错误信息。此外,RAG应用能够在89.2%的情形下正确引用源文件。可见,整合RAG的AI许多时候能够识别正确的指南文件并回答问题,可能会显著减少寻找有关FDA指南文件问题的正确答案所需的时间。但由于FDA指南文件中的信息可能被申办者和FDA依赖用于指导重要的药物开发决策,任何错误信息均可能产生显著的负面影响。未来仍需更多研究,采用各种工程手段来提高答案的准确度。
对AI进行监管:风险评估,禁止与豁免,以及“人-数据-模型”框架
作为全球目前级别最高的AI监管文件,欧盟AI法案对于全球制药行业都有显著的参考意义。可以预期,欧盟药品监管将遵循该法案的理念和各项规定。
该法案首先明确禁止了一系列AI应用,这些应用的共性在于它们可能侵犯个人的基本权利和自由,包括隐私权、自主决策权和非歧视原则。具体来说,这些禁止的应用包括使用潜意识或操纵性技术扭曲个人行为、利用个人脆弱性进行不当影响、基于社会行为或个人特征进行评估或分类导致不公平对待、仅基于个人特征预测犯罪风险、无差别收集面部识别数据、在非医疗或安全情况下推断情绪、基于生物特征数据进行敏感个人信息分类,以及在公共空间进行非必要的实时远程生物识别。这些应用被禁止的原因在于它们可能对个人或群体造成重大伤害,损害其做出知情决策的能力,或在没有充分法律依据的情况下侵犯个人隐私和自由。一些临床入组筛查、临床诊断等临床用AI可能落在这个范畴。不过,法案也有说明,如果用于医疗或安全用途则可豁免。
该法案的另一核心理念在于对AI系统的风险评估和分类,根据其对用户构成的风险程度,实施不同程度的监管。
AI 存在风险,这是不争的事实,且不仅来自所谓“幻觉”导致的差错。在《使用人工智能进行药品研发和供应的益处和风险》 中,WHO对于制药领域中AI应用的挑战和风险进行了深入探讨。WHO识别的风险包括:
1. 偏见。AI技术在药物开发中可能会复制并加剧现有的偏见。这些偏见主要体现在三个方面:训练AI技术的数据集、AI技术开发者的背景,以及技术的部署环境(即情境偏见)。
2. 安全性。如果用于药物开发的算法没有经过潜在错误测试,或者没有验证 它们是否提供例如假阳性或假阴性的建议,患者的安全可能会受到威胁。AI不仅能够识别或设计新的、医学上有益的化合物,还可能被用来发现潜在的有毒化合物,这增加了对生物风险的担忧。
3. 可解释性和透明度。许多算法,如基于人工神经网络的算法,被认为是“黑箱”,即使是开发者也不理解它们的推断和决策。WHO建议所有算法都应在技术使用的环境中进行严格测试,以确保它们满足安全性和有效性标准。
4. 责任和问责制。AI因其不透明性、对人类输入的依赖、互动、自由裁量权、可扩展性、揭示洞察的能力以及软件的复杂性,使得责任分配非常困难。其中一个挑战是AI系统的开发和设计者可能不承担责任,因为AI引导的系统独立于开发者运作,且可能以开发者声称无法预见的方式发展。
5. 隐私和知情同意。药企和技术公司在AI药物开发中使用的数据包括临床试验中的数据,以及来自医院、社交媒体等来源的患者数据,这关系到个人隐私权。
但如此复杂的风险又该如何评估?正如药品的风险最终可以归属到“安全、有效和质量可控”,欧美监管也针对AI风险提出一个简明的模型。欧盟AI法案指出,高风险AI系统的判断标准不仅基于它们对个人的影响,还包括是否对决策产生重大影响。如果AI系统仅执行狭窄程序任务、改善已完成的人类活动结果、检测决策模式而不替代或影响人类评估,或为附件III中列出的用例进行预备性任务,且不进行个人画像分析,则不被视为高风险。企业组织将AI系统投放市场或投入服务前,需记录其评估并可能需要注册。欧盟委员会将提供实施指南和实际案例列表,以明确哪些AI用例属于高风险或非高风险。
7月7日发布的《欧盟GMP附录22人工智能》指南草案 与欧盟AI法案一脉相承。指南草案仅6页,并无太多细节指导,其核心内容首先是明确界定了关键GMP 操作中可以用的AI模型包括:
通过数据训练获得功能的AI/ML模型,而不是通过明确编程实现功能的模型。
静态模型,即在使用过程中不通过纳入新数据来调整其性能的模型。
具有确定性输出的模型,即在给定相同输入时提供相同输出的模型。
以这三个标准,不适用的模型也就可以界定了。指南草案明确不适用于生成式AI和LLM——即当前最火热的ChatGPT和DeepSeek类产品——并明确指出这些模型不应用于关键的GMP应用。即使药企在非关键GMP应用中使用(即对患者安全、产品质量或数据可靠性 没有直接影响),也要始终由具备适当资质和培训 的人员负责确保这些模型的输出适合预期用途,即采用“人在回路”(Human-in-the-Loop, HITL)的方式,并在适用时考虑指南草案中描述的原则。可以看到,这仍然是在贯彻“决策”和“影响”的评估框架,且更加保守,即在GMP领域,AI不应用于影响重大的决策性工作。
这种以“决策”和“影响”评估AI风险的思路同样也能在FDA的指导性文件和指南中找到。2025年1月,FDA 发布《使用人工智能支持药品和生物制品监管决策的考量》 指南草案涵盖广泛的AI技术,并且为企业提出了具体可执行的AI应用评估框架。
该指南以对药品的安全、有效和质量的影响为标准,明确不涵盖以下两类AI应用:药物发现阶段;或用于提高操作效率(如内部工作流程、资源分配、起草/撰写申报资料)。FDA认为这些应用不影响患者安全、药品质量或非临床或临床研究 结果的可靠性。
该指南提出新的概念作为AI监管的基础。可信度(credibility)被定义为通过收集可信度证据而建立的对AI模型在特定使用场景(context of Use, COU)中性能的信任。可信度证据是指任何能够支持AI模型输出在特定COU中可信度的证据。COU则明确了AI模型用于解决特定问题时的具体角色和范围。AI模型的可信度评估活动应与模型风险相称,并针对特定的COU进行调整,以确保AI模型输出在监管决策中的适用性和可靠性。
从COU出发,美国FDA提出了一个“七步走”的可信度评估框架,指导企业评估AI模型的可信度。
第1步:定义感兴趣的问题(question of interest)
第3步:评估AI模型风险。其中,模型风险由模型影响和决策后果两个因素决定。模型影响指AI模型提供的证据相对于其他证据的贡献程度,即是否单独用于某项决策,且无其他信息配合;决策后果描述因错误决策导致的不利结果的重要性,这个因素完全与AI技术独立开来,仅考虑对药品安全、有效和质量的影响。这个双因素模型简明扼要,避开对复杂AI技术的探讨,将评估重点聚焦于决策的过程和影响。
当风险评估后确认需要加以有效的监管,就需要一个框架用于实施监管,也就是制定上述第4步中的“AI模型可信度计划”。FDA在2023年5月发布的《在药品和生物制品研发中使用人工智能和机器学习》 (后在2025年2月略作修订 )文件中提出一个AI监管框架,分为3个核心模块,可供参考:
核心模块1:人类主导的治理、责任和透明度。该模块强调在AI/ML技术规划、开发、使用、修改和停用过程中,人类监督的重要性,以及确保法律和伦理价值的遵守。治理结构要求包括风险管理 计划、透明度和文档记录,以确保关键步骤和决策的追踪和记录,从而增强AI/ML模型的可追溯性和可审计性。
核心模块2:数据的质量、可靠性 和代表性。AI/ML技术对用于训练、测试和验证的数据属性高度敏感,因此,确保数据的质量、完整性和适用性至关重要。该模块涉及对数据偏倚、完整性、隐私和安全性、数据来源、相关性和可复制性的考量。
核心模块3:模型的开发、性能评估、监控和验证。该模块讨论AI/ML模型开发和性能评估的重要性,包括预先规范步骤和模型开发与评估标准的清晰记录。模型风险和可信度是该模块的关键考虑因素,能决定模型在证据总体中的影响力和错误决策的潜在后果。
如何监管一个黑盒:FDA的工具,EMA的实践,以及“周而复始”监管
比上述框架更加具体的,是欧美药监不约而同诉诸于传统的验证范式,将经典理念和流程用于AI,尽管作为一个“黑盒”的AI迥异于以往监管对象,如工艺 、分析方法 、设备 设施 等等。
FDA在《在药品和生物制品研发中使用人工智能和机器学习》 中特别提及美国机械工程师协会(ASME)开发的V&V 40风险信息可信度评估框架。V&V 40最初为评估医疗设备中使用的计算模型的可信度而开发,不特定于AI/ML,但它已被用于医疗器械 和模型引导的药物开发。这一框架给出了在特定用途下AI/ML模型的验证和确认(V&V)过程中基于风险的方法,可以根据模型使用的具体情境,指导所需的证据水平和记录保持程度,其核心在于评估模型的可信度,确保其在实际应用中的可靠性和有效性。通过这一框架,开发者能够识别和量化模型可能带来的风险,并采取相应的缓解措施。这不仅涉及到模型的技术性能,还包括模型在特定临床环境中的实际表现,以及其对患者安全和治疗效果的影响。尽管V&V 40一开始并非专门针对药物开发,但有助于监管部门探索这些标准对药物开发场景的适用性。
上文提到EMA在3月发布了首个基于AI的创新药物开发方法的资格认定意见 。这一名为AIM-NASH的AI工具能够帮助病理学家分析肝脏活检扫描图像,以确定代谢功能障碍相关脂肪性肝炎(MASH,此前称为非酒精性脂肪性肝炎,NASH)的严重程度。EMA从该工具的使用流程,回溯其模型开发、工具验证、临床验证 ,且提供案例分析,全方位阐述监管的考量。AI 工具该如何取信于人,该文件值得致力于AI应用和监管的业界人士仔细研究。
AI的另一个特点是不断的学习与演化,这又不同于传统监管所熟悉的阶段性变更。针对这一点,欧盟AI法案强调了高风险AI系统必须遵守一系列具体要求,这些要求考虑到了系统的预期用途以及AI和相关技术的公认技术状态。法案中提到,高风险AI系统应当建立、实施、记录并维护一个风险管理 系统,这是一个持续迭代的过程,覆盖了整个生命周期 ,需要定期进行系统性审查和更新。这包括识别和分析风险、评估风险、基于数据分析 评估其他可能的风险,并采取适当的风险管理措施来解决这些风险。此外,法案第8条强调了高风险AI系统必须符合这些要求,并且在产品设计中整合必要的测试和报告流程,以确保一致性,避免重复,并最小化额外负担。
这个持续性监管理念与FDA前局长Califf署名 的文章中提到的“周而复始”(recurrent)监管理念是相通的。Califf在文章中指出,AI的性能和效能需要在其实际使用场景中进行持续监控,而不仅仅针对AI本身。这意味着监管范式的转变,从传统的代表性的、基于验证的监管模式,转向了周而复始的乃至连续的监管模式,以适应AI技术的动态性和对环境变化的敏感性。文章还进一步强调AI产品的上市后评估的严格程度应该不亚于其上市前评价。这与传统的药品监管有根本性的不同,因为传统的药品监管通常是基于代表性的上市前临床试验和工艺验证 等活动(尽管AI工具仍然需要通过某种“验证”)。
结语:期待新技术带来新范式
随着AI在制药领域应用的不断拓展,未来药监机构的监管工作将面临更多挑战与机遇。一方面,AI技术的复杂性和动态性要求监管机构不断更新监管理念和方法,以确保其在药品研发、生产、销售等环节的安全性和有效性。另一方面,AI也为监管机构提供了更高效、精准的监管工具,有助于提升监管水平和效率。
可以想见,各国将不断完善相关法律法规和指南,逐步明确AI在药品生命周期 中的应用边界和监管要求。在监管实践中,风险评估和持续监测将成为关键环节。监管机构将基于“影响”和“决策”框架评估AI系统的风险等级,实施差异化的监管策略,确保高风险应用得到严格管控。此外,监管机构还将加强对AI模型的验证和确认,确保其在实际应用中的可靠性和稳定性。
不仅如此,新的技术还会带来新的应用和监管范式,制药AI监管的未来将是一个不断探索和创新的过程。监管机构和药企需通力合作,在保障药品安全和促进技术创新之间找到平衡,为制药行业运用前沿技术实现可持续发展提供有力支持。
识林-实木
识林® 版权所有,未经许可不得转载
岗位必读建议:
研发 :应深入理解模型引导的药物研发理念,合理运用建模与模拟技术,提高研发效率。临床 :需关注模型在临床研究中的应用,如剂量选择和患者亚群体的用药调整。QA :确保模型分析的科学性和可靠性,进行严格的质量控制。注册 :了解模型分析在药物注册中的应用,准备相应的支持性文件。文件适用范围:
要点总结:
模型引导理念 :强调模型引导的药物研发理念应贯穿药物研发全过程,包括非临床到临床研究以及上市后评价。模型分析重要性 :明确模型分析在药物研发决策中的关键作用,提倡合理运用以提高研发效率。数据来源与质量 :强调数据质量对模型分析可靠性的重要性,建议使用充分质量保证的数据。模型评价 :指出模型评价对模型可靠性和稳健性的重要性,建议采用多种方法综合评估。监管考虑 :鼓励运用模型引导的药物研发技术,同时注意模型分析的不确定性,确保科学性和可靠性。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
RA(注册):必读。了解大型语言模型在药品注册中的应用和监管要求。 QA(质量管理):必读。掌握大型语言模型在质量保证过程中的合规性。 研发:必读。确保研发过程中使用的大型语言模型符合监管指南。 临床:必读。了解在临床研究中使用大型语言模型的指导原则。 工作建议:
RA:在注册文件中明确指出大型语言模型的使用情况及其合规性评估。 QA:监控大型语言模型在质量控制中的应用,确保数据的准确性和完整性。 研发:在研发报告中详细记录大型语言模型的使用细节,包括算法的选择和验证过程。 临床:在临床研究报告中包含大型语言模型的使用说明,以及对结果的影响分析。 文件适用范围:
文件要点总结:
合规性要求: 明确指出大型语言模型的使用必须遵守相关的法规和指导原则。数据保护: 强调在使用大型语言模型时,必须确保患者数据的隐私和安全。透明度和可解释性: 要求在使用模型时提供足够的透明度,并对模型的决策过程进行解释。模型验证: 规定了对大型语言模型进行严格验证的必要性,以确保其在监管活动中的可靠性。持续监控: 鼓励对大型语言模型的性能进行持续监控,以适应监管科学的发展。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
RA(注册) :必读,需理解AI在药品和生物制品注册决策中的应用和要求。R&D(研发) :必读,应掌握AI技术在药品和生物制品研发中的应用指导。QA(质量管理) :必读,了解AI技术如何影响质量管理和合规性。工作建议:
RA:评估现有注册流程中AI技术的适用性,确保符合FDA的最新指导。 R&D:在研发过程中考虑AI技术的整合,提高效率和准确性。 QA:监控AI技术在质量管理中的应用,确保符合监管要求。 适用范围:
文件要点:
AI技术应用: 明确了AI技术在药品和生物制品监管决策中的潜在应用,强调了其在提高决策效率和准确性方面的重要性。数据治理: 强调了在使用AI技术时,必须确保数据的质量和治理,以支持有效的监管决策。透明度和可解释性: 特别强调了AI模型的透明度和可解释性,要求企业能够解释AI决策过程。风险管理: 提出了对AI技术相关风险的管理要求,包括对算法偏差和错误结果的识别和控制。监管互动: 鼓励企业与FDA进行沟通,以确保AI技术的应用符合监管要求。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
研发(R&D) :必读。负责模型引导药物研发(MIDD)的规划和实施,确保模型的适用性和评估。注册(Reg) :必读。负责与监管机构沟通,提交MIDD证据,并根据监管要求调整MIDD计划。临床(Clin) :必读。参与MIDD证据的生成,以支持临床决策和药物开发。工作建议:
研发(R&D) :在规划MIDD时,明确模型的目标问题(Question of Interest),并根据模型风险和影响评估结果调整模型评估要求。注册(Reg) :在与监管机构的互动中,使用评估表(assessment table)作为沟通工具,提高透明度,并在规划阶段理解MIDD。临床(Clin) :在临床研究中,利用MIDD证据支持决策,特别是在模型风险和影响较高时,需与监管机构进行早期对齐。适用范围:
文件要点总结:
MIDD证据评估框架: 明确了MIDD证据评估的关键要素,包括目标问题、使用背景、模型影响、错误决策后果、模型风险和模型影响。模型评估: 概述了模型评估的元素,包括验证、验证和适用性评估,并提供了一般性建议。MIDD规划和提交: 提供了模型分析计划(MAP)和模型分析报告(MAR)的建议,以及监管互动和提交的文件要求。监管互动: 鼓励在MIDD规划和证据提交阶段与监管机构进行早期和多学科的互动,特别是在模型风险和/或模型影响预期较高时。技术标准和模型评估: 强调了模型评估应遵循当前接受的标准和/或科学实践,并与模型风险相称。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读指南 QA : 应深入理解人工智能在药品监管中的应用,以提升质量管理的效率和准确性。注册 : 必须熟悉人工智能在注册审批中的应用,优化注册流程。市场 : 需了解人工智能在网络交易监管中的应用,以加强市场监管。研发 : 应掌握人工智能在药品研发数据整理和分析中的应用,提高研发效率。临床 : 应了解人工智能在药物警戒中的应用,优化临床试验监管。文件适用范围 本文适用于化学药、生物制品、疫苗和中药等药品类型,特别针对创新药、仿制药、生物类似药及原料药等注册分类。由NMPA(中国国家药品监督管理局)发布,适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。
文件要点总结 准入审批类应用 形式审查 : 利用人工智能提升注册材料的自动化审查效率,确保材料合规性。日常监管类应用 远程监管 : 结合大数据,通过人工智能模型进行风险预警和数据分析。现场监管 : 人工智能辅助现场检查准备和报告撰写,提高监管效率。服务公众类应用 业务办理及政策咨询 : 人工智能提升客服响应速度,优化公众咨询体验。辅助决策类应用 数据分析与预测 : 人工智能辅助进行市场动态分析,为监管决策提供支持。风险管理 : 人工智能技术用于风险预警和分析,提高监管的前瞻性和精确性。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证) :应深入理解AI模型在GMP环境中的预期用途和相关风险,确保模型的测试、验证和操作符合GMP要求。IT(信息技术) :负责AI模型的技术实施,包括模型训练、测试和操作过程中的技术控制和数据保护。数据科学家 :参与AI模型的选择、训练和验证,确保模型的性能符合预期用途,并进行质量风险管理。研发 :与QA和IT合作,确保AI模型的输入样本空间和预期用途描述准确,包括数据的全面特征和可能的偏差。适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
必读岗位:研发(R&D)、市场(MKT)、注册(RA)、合规(CPL)。 R&D:需关注技术发展与治理章节,确保研发活动符合国家法律法规要求。 MKT:应理解服务规范章节,以确保营销活动不违反社会主义核心价值观。 RA:需全面掌握该办法,指导企业合规注册和运营。 CPL:负责监督企业遵守法律法规,特别是数据安全和个人信息保护。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。