首页
>
资讯
>
使用者付费法案重新授权推进中,FDA该如何促进仿制药竞争?
出自识林
使用者付费法案重新授权推进中,FDA该如何促进仿制药竞争?
2022-07-11
使用者付费法案 重新授权虽经重重博弈 ,但也正在有条不紊地推进 中。促进仿制药竞争无疑是一大热点。
近年来美国各种药品相关政策鼓吹和联邦调查层出不穷,此次重新授权是监管层面改革,降低壁垒不容错过的机会 。The Brookings Institution联合南加州大学的Leonard D. Schaffer Center for Health Policy & Economics(下文中统一称为机构)于6月14日发表了一份白皮书,围绕公民请愿 、复杂仿制药批准以及首仿药 推迟上市三方面提出改进方向。
遏制品牌药公司的花招:别乱“请愿”!
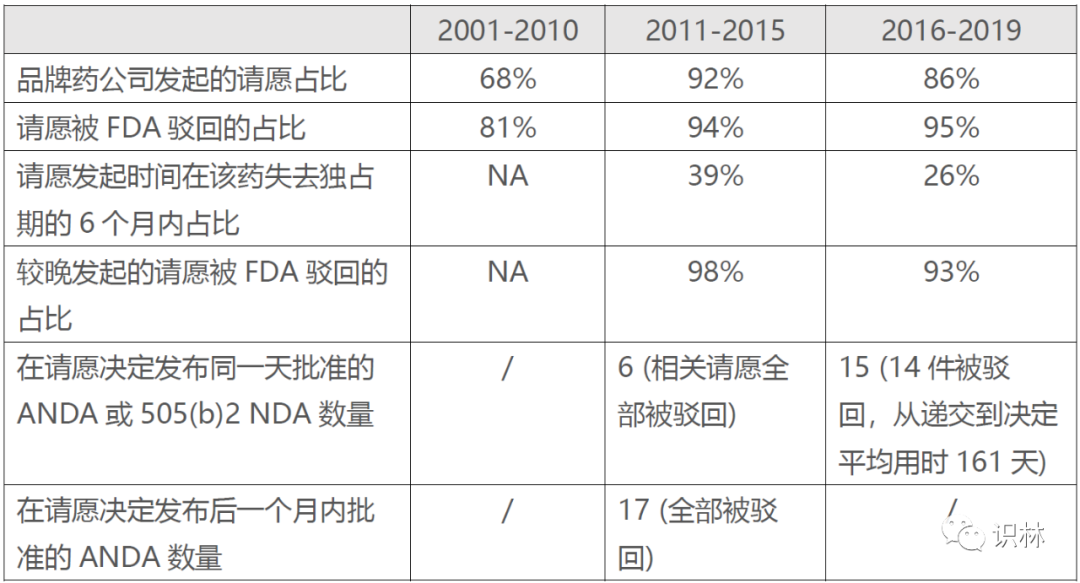
505(q) 公民请愿允许有关各方以保护公众健康为由,引证生物等效性 或安全性问题向FDA请愿,延迟批准待决的仿制药、改良型新药和生物类似药 申请以开展进一步调查。大部分请愿的发起者都是品牌药公司,针对的对象多数是仿制药,其中相当一部分是在临近品牌药失去市场独占期时(6个月内)递交的,拒绝率相当之高,如下表所示。
拒绝率如此之高,再对请愿设限是否有点多此一举了?为了遏止滥用,政府已经通过前几轮UFA重新授权对联邦《食品、药品和化妆品法(FD&CA)》505(q)条 进行修改,赋予FDA立即驳回那些仅为拖延批准的所谓“请愿” 。可是到了实施时,FDA没法一有所怀疑就立刻驳回,还是要对请愿开展评估以证实该请愿未能提出任何有效的科学或监管问题。一旦收到请愿,FDA就需要在150天内作出最终决定,意味着FDA要花费额外的资源和时间,很可能挤占了正常审评的资源。
由于FDA对“拖延”的定义比较宽松,同时报告缺乏细节,比如FDA没有明确哪几个产品受到了影响,仿制药 的批准和请愿驳回等决定常常在同期发布,很难去衡量影响到底几何,尚不清楚FDA对公民请愿本身的评估是否会拖慢仿制药批准。学者们认为拖延的频率可能比FDA报告的要高,也就是说无论结果如何,很有可能只要发起“请愿”,本身就为原研药公司争取时间。
因此机构认为还是要在源头上解决问题,增加威慑力“劝退“恶意请愿。首先FDA应该设定提交请愿的最后期限,根据FDCA(食品、药品和化妆品法案) 要求发起人证明,其请愿所依据的信息为他们所知不超过60天,使FDA有权进行经济或其他形式的处罚,处罚力度也需要加大以达成足够的威慑力。同时FDA审查公民请愿机制的透明度有待加强,应细化FDA的报告,要具体到哪几个请愿以及哪些产品受到了影响,这可能有助于对相关请愿发起公司实施执法活动。
复杂仿制药:提高审评效率和透明度
此处机构聚焦在药械组合产品 ,特别是注射用小分子药品,因为此类产品的数量在不断大幅增长,同时大量已过专利期的产品面临很少或近乎没有仿制药竞争,首仿药平均在过专利期后15年才进入市场。
复杂仿制药的监管难点在于难以建立生物等效性,相较于加拿大和欧盟的复杂仿制药批准情况,美国相对落后 。尽管FDA为了降低复杂仿制药研发的难度制定了特定产品指南,建立一致性仍然非常困难,因为指南出于“商业机密”的考量没有公开具体的配方(formulation)细节。仿制药公司可以通过受控函将配方交由FDA仿制药办公室审查,但是一次只能提交3种配方。如被拒绝,拒绝信中不会提供具体指导说明需要修正什么,因此申请者主要靠猜,一轮下来要花费4个月,而通常在正式敲定配方前要迭代几轮,导致了不确定性很大,效率也低。或者仿制药公司索性转变思路,通过505(b)(2) 途径申请,但是想要替代原研药,还是需要通过公民请愿申请治疗等效性AB评级,而FDA对此类请愿没有规定回复时限,通常要耗费3.5年,获批或收到回复的概率也极低。
“相同性”要求(sameness requirements)中用来增加患者使用安全性的产品特征(比如产品外观,识别标记,包装配置等)通常都受专利保护。虽然FDA容许设计上存在细微差异,但是对细微差异和重大差异的界定比较模糊,差异较大时FDA会要求申请者进行人为因素研究。同时FDA规定患者要能够在没有经过医生/药剂师额外培训的情况下确保足量的API递送并避免用药差错。
针对这些障碍,机构提出:对于药械组合,FDA首先应明确怎样才算重大设计差异,放宽一致性要求,从实质等效(substantial equivalence)转向功能等效(functional equivalence),同时扩大ANDA的标签例外,允许经过医疗保健提供者进行一些适度的培训或使用患者安全工具指导正确的使用。对于注射用复杂仿制药,FDA可以简化配方一致性的审评,通过为被拒绝的申请提供具体的指导来提高透明度,或增加可提出的配方数量。最后FDA应该为505(b)(2) 公民请愿设立一个明确的审查期限(180天),国会也应该针对此建立绩效指标。
激励首仿药尽早上市:胡萝卜加大棒
最后又落到了Hatch-Waxman法案上,识林此前关于专营期的讨论 ,此处就不多赘述背景了,重点放在解决方案上。
针对首仿药公司和原研药公司协商推迟仿制药上市时间,机构建议彻底堵死这条双赢的路,旨在阻止更隐蔽的报酬形式如国外市场销售许可。FDA应明确规定,仿制药和品牌药公司之间的任何专利诉讼和解,只要使得仿制药无法立即进入市场,则应被视为放弃/没收(forfeiture)仿制药的180天专营期 奖励的理由。
针对那些冒着风险上市(at-risk entry)的首仿药 可以制定额外的经济激励措施,适用于以下情况:若法院裁定Paragraph IV 专利挑战不成立,首仿药公司需要向原研药公司支付赔偿金。
白皮书中对以上监管挑战的讨论不禁让人思考,规则指导订得细可能会缺乏灵活度适得其反,较为宽松又会出现空子让人有机可乘,还是要系统地看待问题,不光是平衡各利益相关方,更是要防止规则间相互矛盾造成阻碍。
参考资料
url
作者:识林-梣
识林® 版权所有,未经许可不得转载