2 月 9 日由美国媒体 The Hill 主办、普享药协会(AAM)赞助的“复杂仿制药和处方药格局”网络研讨会上,业界传达了非常重要的信息,尤其是目前针对仿制药使用者付费修正案 III(GDUFA III)的谈判正在试图加快仿制药办公室(OGD)对复杂仿制药的审批程序。
美国企业研究所常驻研究员 Alex Brill 博士研究并发布的一份题为《加速美国复杂仿制药批准可能带来的费用节约》
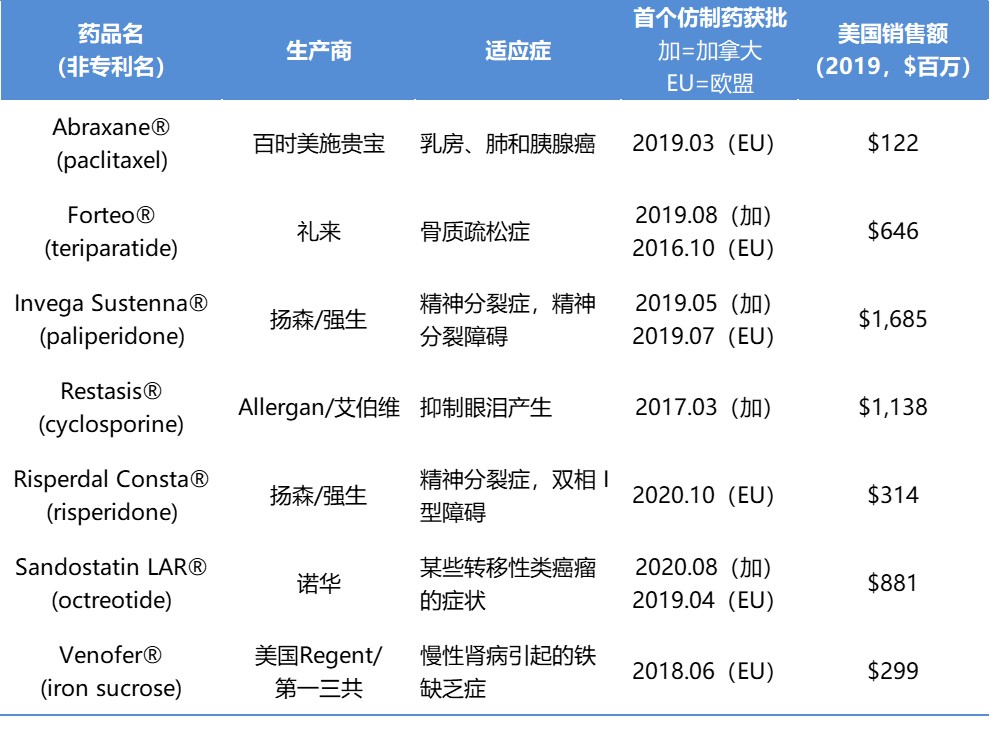
白皮书[1]中得出的结论是,如果现在对美国目前尚未获批的七种复杂仿制药做出批准,那么只这七种复杂仿制药每年就能为患者和美国政府节约 13 亿美元。
Brill 的估算基于下面的假设:鉴于开发困难,大多数复杂产品只能预期有一个或两个竞争对手。他在竞争有限的情况下借鉴了小分子仿制药的经验,认为一两个竞争者的价格折扣可以达到 30% 到 44%。Brill 的分析可用于为在 GDUFA III 谈判或随后所需立法的国会辩论中提议对复杂仿制药程序的变更提供论据。
Brill 在报告中指出,“复杂仿制药代表了美国医疗系统尚未开发的节约机会。FDA 和其他利益相关者已经做出了努力,以建立一个更加强大的复杂仿制药市场,但是到目前为止,这些努力带来的是过程改进而不是成果收益。”在报告中,Brill 列出了七种复杂仿制药,这些产品在加拿大或欧洲已经有仿制药获得批准,且在美国有 ANDA 待审。这些产品 2019 年在美国的总销售额超过 50 亿美元。
成本无疑是 OGD 改善复杂仿制药审批流程的推动力。Brill 博士指出,最重要的事情是 FDA 应当有明确的指南,并且应该为该审批过程提供足够的资源,这在 FDA 审批过程中应该是最高优先级。当被问及关于复杂仿制药的审批应向国会提出的最重要的问题是什么,Brill 表示,“为什么现在需要更长的时间”才能获得批准?
Baeder 指出,“如果我们要继续获得与仿制药相关的节约,我们将不得不弄清楚如何更高效地让复杂仿制药获得批准。我们再也不能等待 44 个月或更长时间来批准复杂仿制药“,她概述了三个对提高 FDA 审批流程的效率最重要的问题:
1. 增加透明度 — FDA 需要更加主动和及时地提供建议。Baeder 认为,FDA 可以通过增加交流来提供更多帮助,例如,在评价过程中出现问题时做出更多努力来通知申办人。
2. 可预测性 — 行业需要知道何时将完成审评,产品何时将接近批准,以准备好上市并满足市场需求。
3. 更好的指南实践 — 为具体产品提供最新信息和 FDA 的思考。她指出,FDA 需要改进与具体产品指南、完全回应函以及提供 FDA 建议的其它方式相关的实践。申办人经常抱怨具体产品指南在没有警示的情况下发生更改,有时会在产品申请已经申报之后,这可能会延迟批准。
Baeder 指出,随着 GDUFA III 重新授权近在咫尺,这是解决围绕复杂仿制药的一些政策问题的最佳时机。共和党众议员 Brett Guthrie 也对此表示赞同,他指出,Hatch-Waxman 法案最初是为简单仿制药设计的,但是随着某些复杂仿制药的出现,法案需要随着时间的推移而发展。Guthrie 表示,FDA 一定不能损害其安全性和有效性评估,但是也需要提高流程效率,以免出现不必要的障碍。申请人参与到 FDA 中的信息越多,申请的质量就会越好,这将取决于 FDA 与行业之间沟通的不断改进。
过敏和哮喘网络总裁兼首席执行官 Tonya Winders 则一语中的。她指出,总体目标是减少审评轮次和沟通次数,而监管者向行业提供明确、没有歧义的建议是解决问题的方法。可靠的 FDA 指南与申请质量相结合,对于减少 FDA 审评轮次至关重要。她指出,仿制 EpiPen 花了十年时间才获得批准。
所有这些讨论要点,如果能转化为 GDUFA III 中的行动计划,则可能有助于加快复杂仿制药的简化新药申请(ANDA)审批流程。但是,必须认识到,如果一件新化学实体(NCE)和/或复杂产品的新药申请(NDA)可以在 10 个月内获得批准,则需要一种更有效的流程来批准在 ANDA 中提交的该产品的仿制产品。
美国仿制药业资深人士 Bob Pollock 在其博客文章中评论指出,“这可能需要与潜在申办人举行更多的会议,并确保 FDA 工作人员在收到首个 ANDA 之前完全了解如何将参照上市药品与仿制药进行比较。尽管 FDA 在这方面正在追赶,但是在过去,他们主要依赖在申请提交到 FDA 之后与 ANDA 申请人一起学习。对于这些复杂产品以及目前没有竞争产品的仿制药,必须改善沟通。从 FDA 和行业角度来看,行业中应该有其它沟通接触点来解决在申请审评过程中发现的问题。我认为,目前的受控函(CC)流程显然不足以澄清申报要求,并且如果 OGD 继续对生物等效性建议和参照标准请求做出零碎回应(或根本不响应),受控函未来也无法澄清申报要求。当申请人想将市场上没有 NDA 或 ANDA 产品的情况下将旧的 NDA 产品重新推向市场时,尤其如此。一些询问 BE 建议的 CC 函持续了两年半之久,没有任何回复或针对具体产品的指南发布。GDUFA III 也许可以解决其中一些问题 , 但是当需要更深入的对话来解决问题时,FDA 必须寻求在 CC 函程序之外的互动方式。在这种情况下,FDA 应与业界联系 , 以就复杂或异常情况或问题进行对话。拒绝 CC 请求或者让其长时间搁置都不是解决问题的方法。我充满希望,但是怀疑这些政策问题和进展能否全部纳入 GDUFA III 的重新授权之中。最后 , 至关重要的一件事是,在就目标函的内容达成一致之前,应确保 FDA 和业界对目标函的条款具有相同的理解。”
[1] Alex Brill, Potential Savings from Accelerating US Approval of Complex Generics, 2021/02.