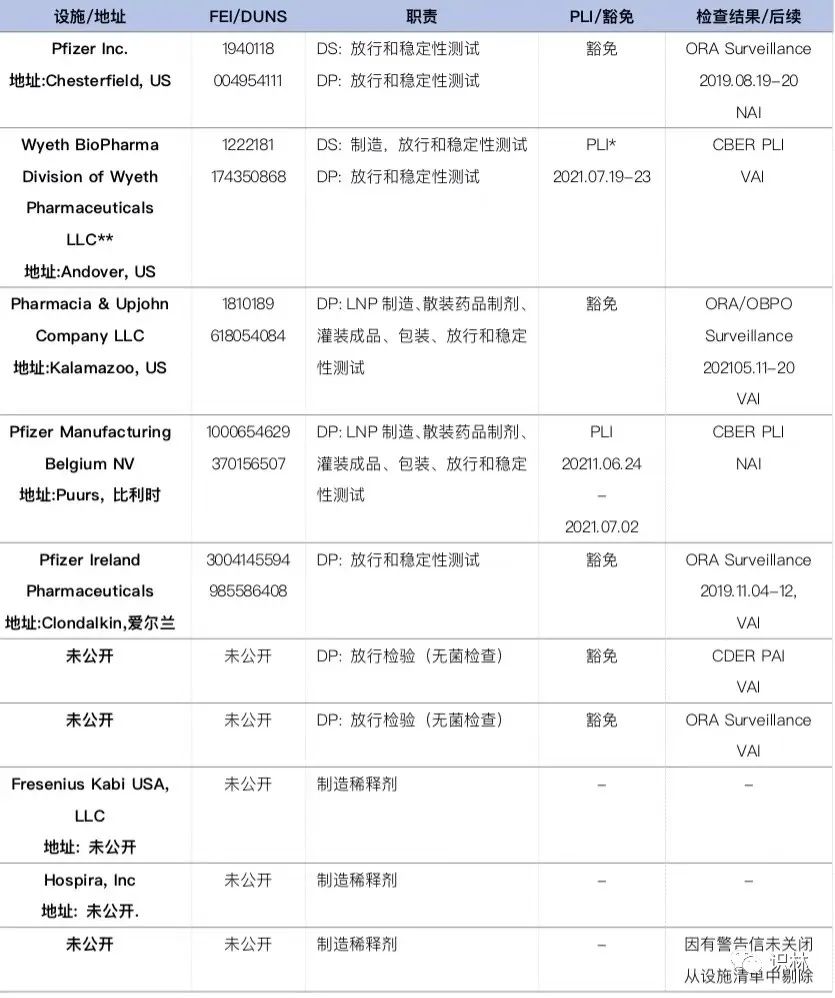
Comirnaty BLA 及补充申请文件中涉及的与疫苗制造相关的设施分布在全球多个国家或地区,下表列出了参与 Comornaty 制造的设施,并标明了 FDA 基于审评和风险决策开展的 PLI 检查、监督检查活动,以及检查结果分类情况。2021年7月19日至23日,CBER 对 Pfizer Andover 的许可前检查(PLI),签发了13条观察项,最终所有问题得到解决,检查被归类为自愿行动指示(VAI)。其他场地或豁免 PLI,或检查结果为 VAI 和 NAI;或 ORA 进行的监督检查,检查结果为 VAI 或 NAI。另外,一稀释剂制造设施因有未关闭的警告信,致使被迫在设施清单中剔除。
* DS: Drug Substance, 原液;DP: Drug Product, 制剂;PLI: Pre-license inspection,许可前检查。
**Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC,下称 Pfizer Andover;其他简称同为 Pfizer+城市。
稀释剂制造设施因存在警告信未关闭,致使在申请文件设施清单中剔除
使用 COMIRNATY™ 疫苗时,需要在接种之前用 0.9% 氯化钠注射液(USP 稀释剂)进行稀释。辉瑞在初始 BLA 文件模块3中没有提供相关稀释剂供应商的信息,后续在补正文件125742/0.47的制造商清单中增加了3个稀释剂制造商。在审评过程中,CMC 审评人员发现其中之一的稀释剂制造商存在未关闭的警告信,仍处于官方指示行动状态(OAI)。FDA 不会批准该设施以支持 BLA 的批准。通过 IR 沟通,递交补正文件125742/0.56,辉瑞从 BLA 中删除了对生产商设施的每一处引用,产品最终在一周后获得批准。
在 EUA 请求时,申办方报告称,在 DP 制造过程的目检步骤中,在多个制造场地的多个批次中观察到不同程度的白色颗粒,涉及有两个灌装完成场地等(其他场地信息被遮挡)。经调查确定这些可见异物属于内源性异物(Intrinsic particulates),具体组成信息被遮挡。[Upon investigation by xxx, the visible particles were identified to be composed of xxx, and thus intrinsic to the product.] 内源性异物是指来自于生产设备、产品处方或容器系统的异物。基于这些调查,辉瑞修订了可见异物(外观)标准,从临床批次的“基本不含可见颗粒(essentially free from visible particulates)”修改为用于紧急供应/商业批次的“可能含有白色至灰白色不透明的无定形颗粒(may contain white to off-white opaque, amorphous particles for emergency supply/commercial lots.)”。文件还指出,进一步的研究证明这些可见颗粒并不对产品质量(如 RNA 含量和 xxx 等)造成影响。