首页
>
资讯
>
FDA 延长亚硝胺相关杂质限度实施提交期限
出自识林
FDA 延长亚硝胺相关杂质限度实施提交期限
2025-06-27
6月23日,FDA在其“亚硝胺杂质可接受摄入量限度 ”页面上发布消息,鉴于药企在应对亚硝胺 药物成分相关杂质(NDSRIs)限度实施工作时面临挑战,决定延长提交的期限,为药企提供了更充裕的时间来完善相关限度实施与控制策略 。
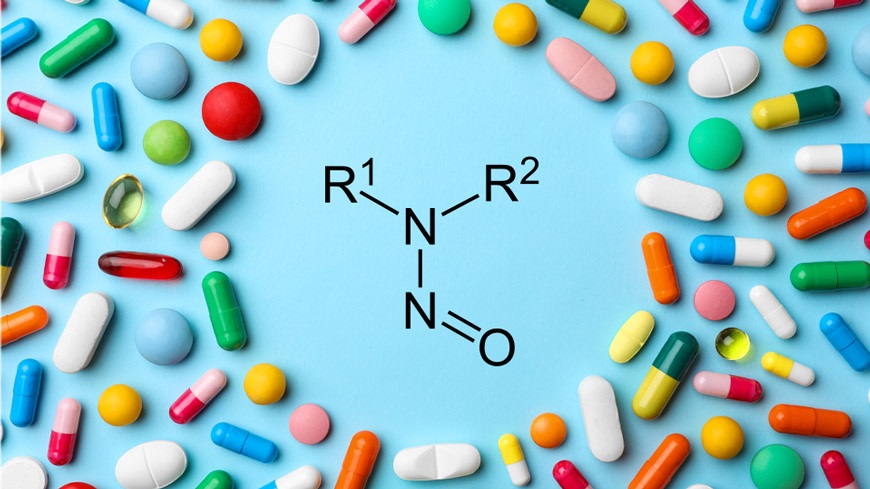
亚硝胺类杂质 问题最早可追溯至2018年 ,当时在缬沙坦(valsartan)类药品中被发现含有亚硝胺类杂质,迅速引起全球监管机构的高度关注。此后,监管机构持续加强对药品中亚硝胺类杂质的监管力度,药企也面临着更为严格的限度实施与控制要求。
此前,FDA规定药企需在2025年8月1日前完成对存在亚硝胺类杂质风险药品的确认性检测,并提交必要的变更申请。但众多药企反馈,由于不同药品的限度实施与控制策略 复杂多样,如增加检测项目或进行药品处方变更等,原定的时间框架难以达成。一些药企可能需要额外时间来优化检测方法,另一些药企则可能在寻找合适的替代原料或改进生产工艺方面遇到困难,从而影响了整体进度。
鉴于药企面临的实际困难,FDA对于无法在原定8月1日截止日期前完成限度实施提交的药企,将给予额外时间。在此期间,药企需在新药申请(NDAs) 、简化新药申请(ANDAs) 以及生物制品许可申请(BLAs) 的年度报告 中提交进展更新,内容需包含:
分析的产品批次信息以及分析日期相对于生产日期的时间关系;
药品中NDSRIs的确认性检测结果(以纳克/天或ppm表示);
当有必要控制时,描述对已识别NDSRIs进行控制的尝试;
FDA此前已发布多份指南,为药企应对亚硝胺类杂质问题提供技术指导。2023年8月发布的《亚硝胺药物成分相关杂质的推荐可接受摄入量限度》 (Recommended Acceptable Intake Limits for Nitrosamine Drug Substance-Related Impurities),基于预测的致癌潜力分类,为NDSRIs的可接受摄入量 提供了推荐标准。2024年9月发布的《人用药中亚硝胺杂质控制》 (Control of Nitrosamine Impurities in Human Drugs)则进一步明确了亚硝胺类杂质的两类划分:小分子亚硝胺和NDSRIs,并详细阐述了相应的控制要求。
截止日前,FDA已在页面“亚硝胺杂质可接受摄入量限度”中罗列了大量亚硝胺杂质的推荐限度,三张表格合计达到301个,此外还包含14个检测方法。
EMA则在其问答指南《MAH/申请人问答:关于人用药中的亚硝胺杂质(条款5(3)转介)的 CHMP 意见》 的附件1 表格中罗列了总计218个亚硝胺杂质的推荐限度。
识林-实木
识林® 版权所有,未经许可不得转载
适用岗位:
QA(质量保证):负责确保药品生产和质量控制符合EMA的N-亚硝胺可接受摄入量(AIs)要求。 注册(Regulatory Affairs):需了解并应用这些AIs以支持药品注册和合规性。 研发(R&D):在药物开发过程中考虑这些AIs,以避免或控制N-亚硝胺的生成。 工作建议:
QA:监控生产过程,确保N-亚硝胺水平不超过规定的AIs,并在必要时进行风险评估。 注册:在药品注册文件中包含关于N-亚硝胺控制的相关信息,并确保符合EMA的最新要求。 研发:在药物设计和工艺开发阶段,评估和控制N-亚硝胺的风险,以满足EMA的AIs要求。 适用范围:
要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议 MAH/申请人 :负责确保药品中亚硝胺含量控制在规定限度内,设计或调整生产工艺以预防亚硝胺形成,并在必要时提交上市许可变更。QA :监督药品生产过程,确保符合GMP规范,审核亚硝胺风险评估和控制策略。研发 :在药品开发阶段考虑亚硝胺风险,参与制定和优化生产工艺。注册 :了解法规要求,准备和更新药品注册文件,包括亚硝胺风险评估和控制措施。文件适用范围 本文适用于所有人用药(化学药和生物制品),包括创新药、仿制药、生物类似药和原料药。适用于在欧盟上市的药品,由EMA发布,适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业。
文件要点总结 亚硝胺风险评估 :所有人用药必须进行亚硝胺风险评估,无论市场状态或产品类型。审查呼吁 :针对化学合成API和生物API的药品,要求MAH进行风险评估、确认性测试和风险缓解措施。风险缓解措施 :MAH需设计生产工艺和控制措施,以预防或减少API和成品药中N-亚硝胺的存在。限度设定 :根据ICH M7(R2)原则,为亚硝胺设定特定的可接受摄入量(AI)限度。监管要求 :欧盟监管机构与国际伙伴合作,协调监管要求,确保药品质量和安全。以上仅为部分要点,请阅读原文,深入理解监管要求。