首页
>
资讯
>
FDA 发布 ICH Q12 实施考量指南,详述如何实施既定条件并提供示例
出自识林
FDA 发布 ICH Q12 实施考量指南,详述如何实施既定条件并提供示例
2021-05-26
美国 FDA 于 5 月 20 日发布《ICH Q12:FDA 监管产品的实施考量》指南草案 ,向制药商解释了如何在各类申请,包括新药申请(NDA) 、生物制品许可申请(BLA) 以及简化新药申请(ANDA) 中提交既定条件(Established Conditions, EC),以及对 EC 的变更报告类别如何确定,以帮助制药商实施 ICH Q12 。
ICH Q12 指南于 2019 年 11 月定稿,旨在促进对新的和已上市药品和原料药 的批准后化学、生产和控制(CMC) 变更 的管理,推进制药领域的创新和持续改进,并加强质量保证和可靠的产品供应。EC 是指南的核心概念,定义为“对于确保产品质量是必须的、具有法律约束力的信息(或被批准的事项)”,对于 EC 的任何变更都需要提交给监管机构。【ICH Q12 指南定稿,更多新指南雏形初现 2019/12/04】
FDA 表示,实施指南草案是对 ICH Q12 的补充,阐明了“如何在美国监管体系内实施 ICH Q12 工具和推动力。”ICH Q12 报告人、FDA 药品质量办公室(OPQ)药品质量政策办公室主任 Ashley Boam 在 5 月 14 日美国 FDA 与加拿大卫生部联合举办的 ICH 区域公开会议上介绍了 ICH Q12 在 FDA 的实施情况。Q12 指南取代了 FDA 2015 年发布的《既定条件:已获批药品、生物制品需要报告的 CMC 变更》 指南草案。
Boam 表示,FDA 从 2019 年启动的既定条件试点计划中获得的经验指导了实施指南的制定【FDA 启动有关药品质量的“既定条件”试点计划 2019/02/19】 。另外 FDA 药品审评与研究中心(CDER)还正在起草有关 ICH Q12 实施的政策程序手册(MAPP)。现有 FDA 指南和法规,包括详述一系列广泛批准后变更的放大和批准后变更(Scale-Up and Postapproval Changes,SUPAC)指南,均与 ICH Q12 保持一致。
实施指南
ICH 关于 EC 的概念也与 FDA 在 21 CFR 314.70(a)(1)(i) 、314.97(a) 和 601.12(a)(1) 中的规定“一致”。尽管这些法规并未指定 EC 的构成要素,但这些法规条款建立了基于风险的系统来报告变更。实施指南表示,ICH Q12“帮助申请人 弄清了在其申请中有关产品、制造工艺 、设施 和设备以及控制策略 的哪些元素被认为是 EC,需要报告变更。”
提交原始 NDA、BLA 或 ANDA 时,申办人应包括提议的具体 EC 以及没有提议的具体EC。对于补充申请 ,申请人应在封面函以及电子通用技术文件的 3.2.R 节列出 EC。申请人应为每个 EC 提出报告类别;类别包括需事先批准的补充申请(PAS)、30 天生效变更补充申请(CBE-30)、立即生效变更补充申请(CBE)或年度报告。
实施指南还说明了对于在美国报告批准后变更如何解释 ICH 术语。例如,Q12 概述了批准后变更的两个类别:事先批准(prior approval)和通知(notification)。而在美国监管体系中,事先批准表示 PAS,中等程度通知表示 CBE-30 补充申请,低程度通知表示 CBE-0 补充申请或年度报告。
文件示例
另外,实施指南还包括三个附录:
附录A 具有器械组成部分的组合产品的既定条件
附录B 对器械组成部分确立既定条件和报告类别的决策树
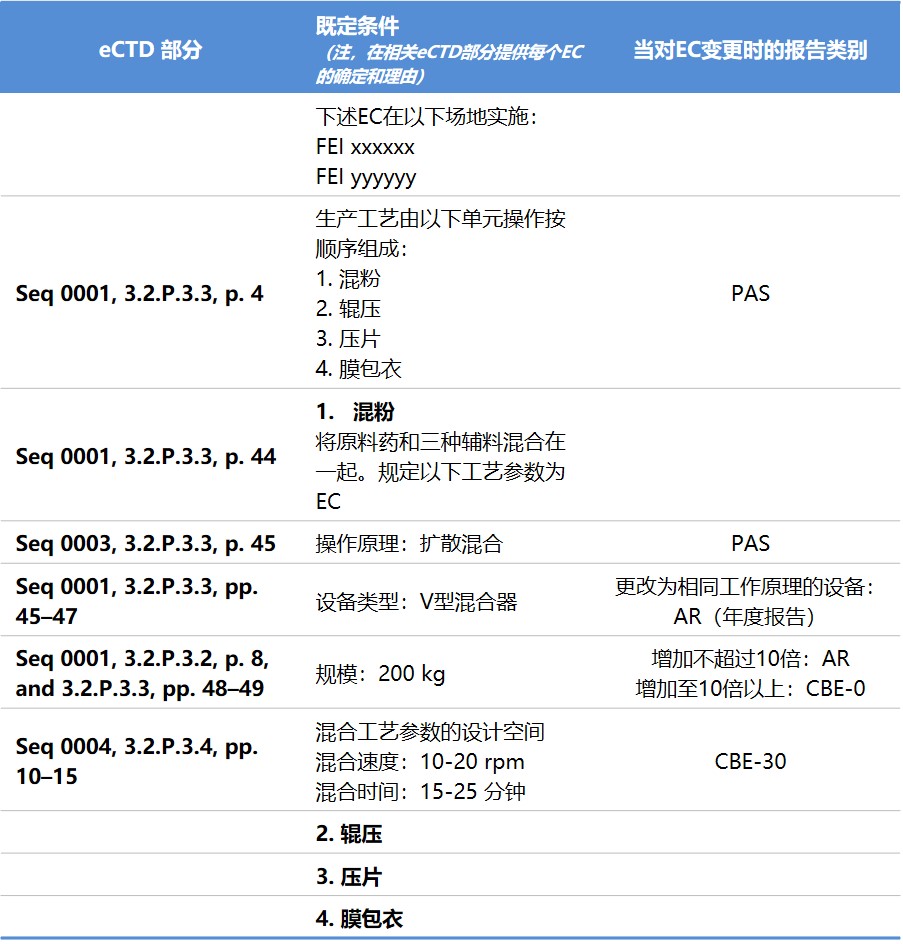
附录C 生命周期管理文件示例
下面是“附录C 生命周期管理文件示例”,在示例中,如果申请人的拟议遵循 FDA 法规和有关对具体既定条件的变更指南中的建议,则报告类别为空白。
作者:识林-蓝杉® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
适用岗位:
QA(质量保证) :必读。建议深入理解ICH Q12指南中关于已建立条件(ECs)的识别和管理,以及如何与FDA的监管框架相结合。注册 :必读。建议熟悉ICH Q12指南中关于药品生命周期管理的监管工具和原则,以便在注册申请中正确应用。研发 :必读。建议了解ICH Q12指南对于药品研发过程中CMC变更管理的影响,特别是在提出ECs时的科学依据。生产 :必读。建议掌握ICH Q12指南中关于生产过程中变更管理的要求,以及如何在生产质量体系(PQS)中实施。适用范围:
要点总结:
已建立条件(ECs)的管理 :强调了在提交原始NDA、BLA或ANDA时,申请人应明确提出特定的ECs及其变更的报告类别。变更管理协议 :提出了通过补充申请或后批准变更管理协议(PACMP)来添加、消除或修改已批准的ECs。药品生命周期管理文件(PLCM) :建议在eCTD部分3.2.R中以表格形式提供PLCM文件,包括提出的ECs、变更报告类别、可比性协议列表和后批准CMC承诺。药品质量体系(PQS) :强调了PQS在支持ICH Q12工具使用中的重要性,并指出FDA将通过常规检查和其他信息来评估PQS的有效性。监管评估与检查的关系 :明确了ICH Q12工具的使用不会改变FDA对申请信息的评估或设施检查的流程。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
QA(质量保证) :应全面理解ICH Q12指南,确保质量体系与监管要求一致,指导产品生命周期管理。注册部门 :需熟悉ICH Q12指南,以便在药品注册过程中有效应用,确保申报材料符合监管要求。研发部门 :应了解ICH Q12指南中关于药品开发阶段的技术和监管考虑,以促进创新和持续改进。生产部门 :需掌握ICH Q12指南,特别是在已建立条件(ECs)和变更管理协议(PACMP)方面的要求。文件适用范围:
文件要点总结:
变更管理框架 :提供了一个框架,以更可预测和高效的方式管理批准后CMC变更。已建立条件(ECs) :明确了MAH与监管机构之间的共识,规定了确保产品质量的要素,以及变更时需要进行监管沟通的条件。变更管理协议(PACMP) :提供了一种监管工具,允许MAH与监管机构就变更所需的信息和监管提交的类型达成预先协议。产品生命周期管理(PLCM)文件 :作为ECs和变更报告类别的中央存储库,捕捉商业阶段产品如何被管理。药品质量体系(PQS)与变更管理 :强调了PQS在管理供应链和产品生命周期中的变更管理的重要性。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议:
QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。 QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。