首页
>
资讯
>
FDA 前仿制药负责人敦促企业保障申报材料质量
出自识林
2019-03-25
美国 FDA 仿制药办公室刚刚退休的主任 Kathleen "Cook" Uhl 强调申请人在 ANDA 提交和审评过程中采取“缓慢”和仔细的方法实际上会获得更快批准。
去年九月她在普享药协会(AAM,原仿制药协会)主办的“GRx+Biosims”会议上的演讲中表示将申请匆匆提交到 FDA 往往适得其反,对申办人 和 FDA 都有害无益。
Uhl 已经于今年 2 月份退休,她在这次会议上的报告带有告别演说的意味。她提供了关于仿制药 计划进展以及仍待解决的挑战的全景介绍,这其中就包括关于行业可以如何帮助确保申请尽可能快地通过审批流程的宝贵见解。
仿制药计划达到“稳态”
Uhl 在开始她的演讲时回顾了 FDA 仿制药计划的现状,以及一些数据和分析。她介绍了仿制药计划在满足和“超越”所有 GDUFA 目标以及在申请“输入和输出”中达到“稳态”等方面取得的成功。她表示仿制药计划现在“处于非常良好的状态”,并且可能是自创立以来“最好的状态。”
Uhl 随后讨论了仿制药计划面临的持续挑战,以及行业可以采取哪些措施来帮助解决这些挑战。
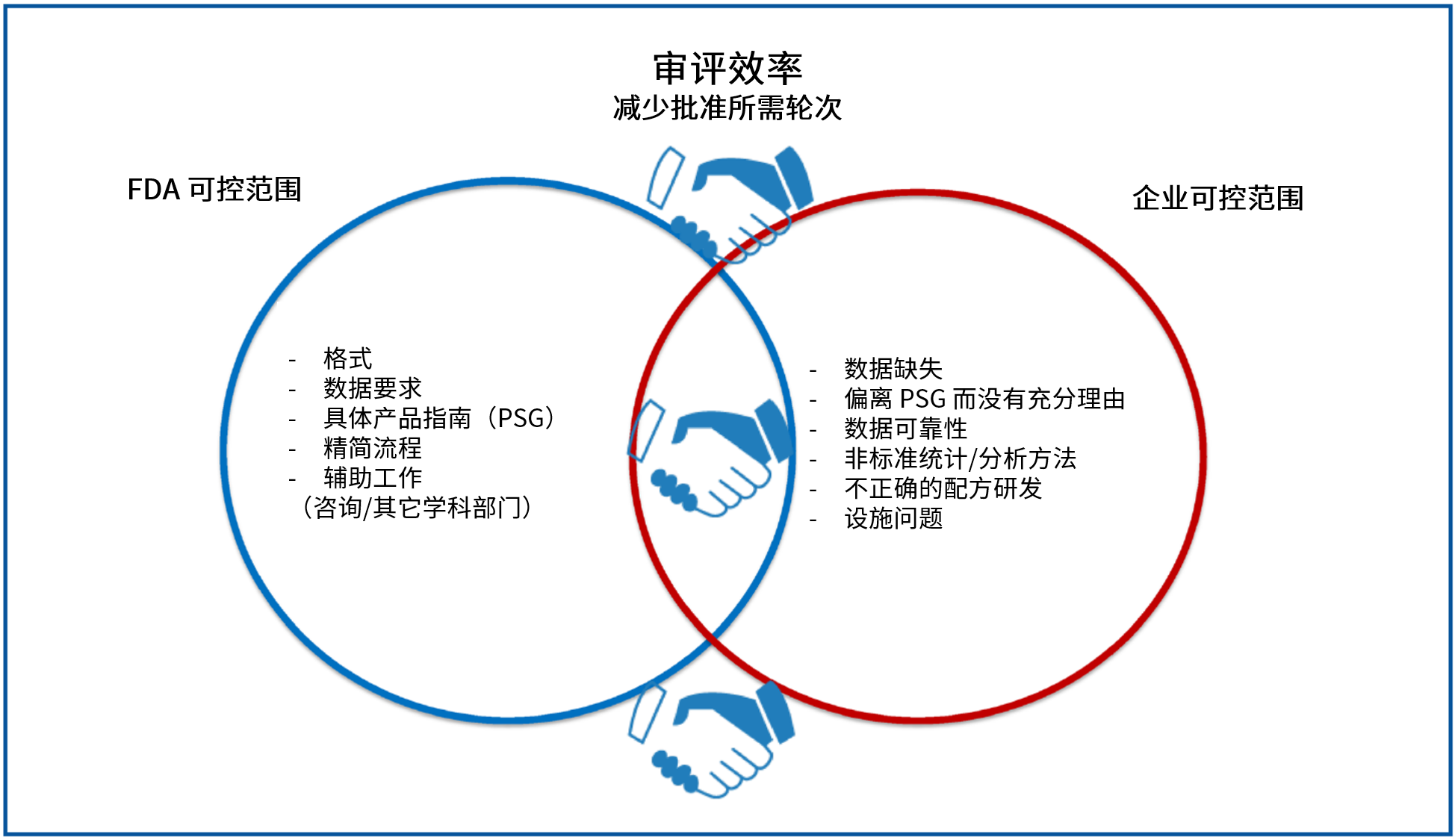
为避免令企业苦恼的审批延迟的发生,Uhl 强烈敦促申请人花费额外的时间与精力来理解和解决:
在研发和编写申请的过程中,充分利用提交前会议和受控函 (controlled correspondence, CC)提供的机会需要做哪些准备
确保申请材料完整并且讲述一个清晰、连贯的故事需要哪些方面的准备
收到拒绝接收(RTR) 认定的申请存在的缺点
在信息请求(IR)中实际要求提供些什么
完全回应函 (CR)通知中描述的缺陷
Uhl 强调,OGD 认识到 RTR 问题对申请人的重要性,花大力气制定了有关 RTR 以及如何提高申报质量的指南和 MAPP,并且希望企业将这些建议牢记在心。她提到导致 RTR 的三大原因是:稳定性 研究不充分;溶出研究不充分以及杂质 问题。她重点提到有临床终点 研究的 ANDA ,这类申请需要花费大量工作、时间和金钱。因申请缺陷而导致 RTR 的话代价更大。她列举了这类申请的 RTR 原因有:未能提供数据集,信息缺失,临床终点 BE 研究未能显示生物等效性(BE)等等。其中绝大多数 RTR 原因是申请中与临床研究有关的数据集和相关信息的缺失。对于这类申请的底线就是,提交包括所有数据集在内的完整临床研究报告。
关于收到 RTR 的申请在重新提交后整个审评过程中仍然存在更多问题,她告诫企业不要“仅仅修订一两个方面的问题”后就重新提交申请以期望申请尽快回到审评轨道,这往往会弄巧成拙。她敦促申请人“请放慢速度。时间并非一切 — 质量至关重要。在初始提交或重新提交之前,真的要对申请开展质量保证 和质量控制 。”
提高首轮批准率需要企业帮助
在回顾首轮批准数据时,她指出,在 2017 年 9 月份结束的 GDUFA I 第五年,首轮批准率实际上比前两年有所下降,仅有 10% 左右。她表示,首轮批准率的下降与 2017 财年使用者费目标行动日期从 15 个月降到 10 个月有关。
她表示,“审评更快并不意味着你将获得更多的首轮批准。申请在 FDA 的时间越短,来回纠正缺陷的机会就越少。”虽然申请人一直在推动缩短审评时间,但有些事情是需要企业自己做的,以帮助提高首轮批准数量。她表示,企业在处理生物等效性方面做得很好,大约 70% 的 BE 材料在首轮审评中被认为是充分的。然而,标签比较成问题,几乎每件提交都有标签问题,需要审评部分发送学科审评函 或信息请求来获得关于标签的内容。因为标签是动态的,参照药品(RLD) 、USP以及专利和专营权 等等在审评过程中都可能发生变化。
Uhl 接下来就企业可以做些什么来帮助实现首轮批准目标提出了更具体的建议。她敦促申请人“讲述你的故事,以便我们理解。使用申请材料证明你了解你的产品,并且你也了解 RLD 的关键属性,并在申请中收录这方面的知识。”她告诫说,如果公司采取“先提交,后修改”的策略,那么将不会改善首轮批准率。
整理:识林-椒® 版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
参考资料 Retiring FDA Office of Generic Drugs Director Uhl Cautions Industry that Haste Makes Waste in Application Approval Timelines