|
首页
>
资讯
>
FDA人数“翻倍”历程:权、责、人的匹配
出自识林
FDA人数“翻倍”历程:权、责、人的匹配
2025-03-28
在3月6日举行的参议院听证会上,候任FDA局长Marty Makary(本文发稿时已获参议院正式任命)被问及近期的全球业界关注的FDA裁员问题。尽管Makary没有具体说明他将如何重新调整,但他倾向于同意对FDA进行整体人员精简。他还提及从2007年到现在,FDA的雇员“翻倍”。
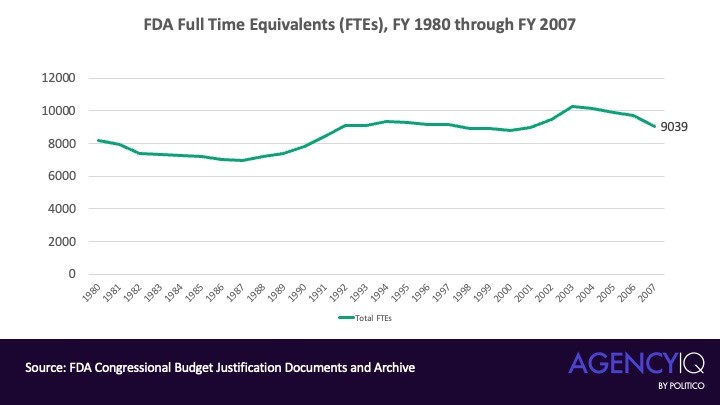
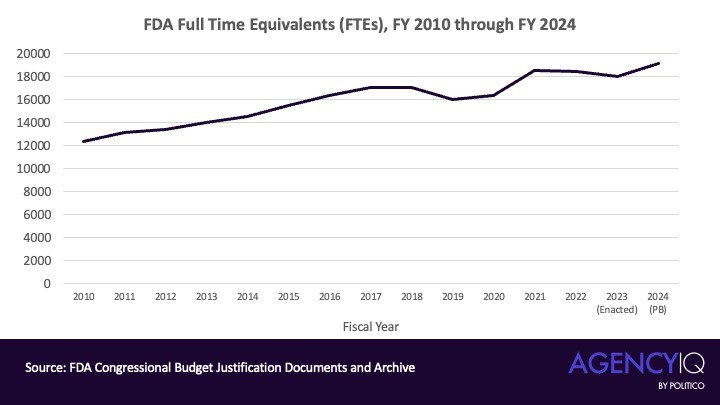
美媒Politico针对这个提法,基于政府官方的预算文件进行了一系列调研,显示FDA的人员规模在2007年时为9,039个全职等效岗位(FTE),而到了2024财年,这一数字已飙升至19,116个FTE,确实翻了一番。
从政府更迭角度:奥巴马时期迅速增长
如上图所示,从1980直到2007,FDA的人员规模并无显著增长。但从2007年开始,尤其是奥巴马的两届任期(2009年1月至2017年1月)期间,FDA的人员规模呈现出稳定的年度增长,年增长率在1.73%到6%之间。从2010财年到2017财年,FDA共增加了4,641个FTE,整体增长率达到37.5%(见下图)。
进入特朗普第一任期后,趋势稍有变化。在2017财年,FDA的人员规模保持在约17,000人,相对稳定。但在2019财年,FDA的人员规模出现了约6.5%的下降。
然而,2020财年新冠疫情的爆发彻底改变了这一趋势。在2021财年,FDA的FTE数量增长了超过11%,达到18,501人。这一增长主要是为了应对新冠疫情带来的公共卫生紧急情况,FDA需要更多的人力来加速疫苗和治疗药物的审批流程,确保这些关键产品尽快上市,以应对全球性的健康危机。
在拜登时期,FDA的人员规模保持了相对稳定,仅在2021财年之后增长了约3.5%。
从业界最为关注的审评部门角度看,FDA的三个主要医疗产品中心——药品审评与研究中心(CDER)、生物制品审评与研究中心(CBER)和医疗器械与放射健康中心(CDRH)——在2010财年至2024财年期间的人员规模分别增长了86%、20%和55%。这显示出FDA在不同领域的监管需求和工作重点的变化。
接着,就是如今特朗普2.0时期,FDA的人员规模再次面临巨大挑战。一系列裁员举措的背后,是特朗普政府对联邦雇员规模的整体削减政策。
从法规政策角度:权力、责任和人员数量的匹配
但政府更迭只是表象,人员增长的真正驱动力,是法规赋予FDA的权力和责任逐年增加。
过去15至20年间,美国通过了一系列具有里程碑意义的立法,显著扩大了FDA的监管和项目范围,也为其人员规模的增长奠定了基础。
这些立法包括但不限于:
- 2009年:《家庭吸烟预防与烟草控制法案》(Family Smoking Prevention and Tobacco Control Act)。该法案授予FDA监管烟草产品制造、分销和营销的新权力。这一立法标志着FDA在公共卫生领域的职责扩展,为其增加了新的工作内容和人员需求。
- 2011年:《FDA食品安全现代化法案》(FDA Food Safety and Modernization Act)。该法案赋予FDA新的食品安全标准执法权力,并要求FDA与州和地方当局共同建立一个综合的国家食品安全系统。这一立法显著增加了FDA在食品安全领域的监管职责,推动了人员规模的增长。
- 2013年:《大流行和所有危害准备再授权法案》(Pandemic and All-Hazards Preparedness Reauthorization Act)。该法案完善了FDA关于紧急使用授权的权力,赋予其延长货架期和豁免某些制造要求的新权力,并扩大了医疗对策的权力。
- 2020年:《新冠病毒援助、救济和经济安全法案》(Coronavirus Aid, Relief, and Economic Security Act)。该法案赋予FDA新的权力,以增强药品短缺的识别和缓解能力,并收取非处方产品的用户费用。
- 2022年:《化妆品监管现代化法案》(Modernization of Cosmetics Regulation Act of 2022)。该法案扩大了FDA通过制定现行良好生产规范法规监管化妆品的权力。
这其中尤其关键的,是一系列使用者付费的扩展,为人员增长提供最为重要的资金支持。但这些资金一样与权责挂钩,即通过与行业的协议,明确了FDA的招聘目标和绩效指标,要求FDA在特定时间内完成审批任务。
FDA并非美国政府成本高企主因
FDA的人员真的过多吗?支持FDA的政策法规专家Grossman在其博客FDA Matters中算了一笔账,用数据为FDA背书。他指出FDA的人员规模和预算支出并非美国政府成本高企的主要因素,而与此同时,FDA在保障公众健康安全方面发挥着不可替代的作用。
美国联邦政府的雇员规模庞大,2023年约有230万名全职雇员(不包括邮政服务)。其中,国防和安全相关机构占据了70%的联邦雇员。人员最多的三个部门分别是国防部(775,100人)、退伍军人事务部(433,700人)和国土安全部(212,000人)。
相比之下,FDA在2023年的人员规模仅为18,500人。这意味着每125名联邦雇员中,只有1人是FDA的员工。
尽管人员规模较小,但FDA的职责却极为重要。FDA的预算约为60多亿美元,负责监管的产品占美国消费者支出的21%。在全球范围内,FDA监管着价值3.9万亿美元的食品、药品等产品。这意味着平均每位FDA员工需要监管约近2.1亿美元价值的产品。
Grossman指出,当前美国联邦政府面临着巨大的预算赤字。2024财年,联邦政府支出6.75万亿美元,收入4.92万亿美元,年度赤字达到1.83万亿美元。特朗普政府认定裁员势在必行,但与国防部、退伍军人事务部和国土安全部等人员众多的部门相比,FDA的人员规模相对较小,而其监管工作不但涉及巨大的市场价值,还直接关系到公众健康和安全,任何人员削减都可能对药品审批、食品安全和医疗产品监管产生负面影响。
识林-实木
识林®版权所有,未经许可不得转载
岗位必读建议: - 研发(R&D):了解加速药品开发流程的新规定,确保研发项目符合新法规要求。
- 注册(Regulatory Affairs):掌握法规对药品注册流程的影响,优化注册策略。
- 临床(Clinical):关注临床试验设计的新指导原则,确保试验合规性。
文件适用范围:
本文适用于美国境内的化学药、生物制品、疫苗等药品类型,包括创新药、仿制药、生物类似药及原料药。主要面向Biotech、大型药企、跨国药企等企业类别。 文件要点总结: - 加速药品开发流程:强调了加速药品从发现到上市的整个流程,以促进21世纪医疗创新。
- 临床试验现代化:提出了对临床试验设计的现代化要求,以提高试验效率和患者参与度。
- 个性化医疗推进:鼓励发展个性化医疗方法,包括精准医疗和基因疗法。
- 数据共享与隐私保护:规定了数据共享机制,同时强调了患者数据的隐私保护。
- 监管框架更新:明确了对FDA监管框架的更新,以适应新兴医疗技术和产品。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位: - QA(质量保证):确保药品生产和供应链符合FDA规定。
- 注册:负责药品注册流程,确保符合新法规要求。
- 供应链管理:监督药品供应链安全,实施新标准。
工作建议: - QA:审查生产流程,确保符合FDA对药品质量的新规定。
- 注册:更新注册资料,反映Drug Quality and Security Act的新要求。
- 供应链管理:制定或更新供应链安全计划,确保符合FDA标准。
适用范围:
本文适用于美国境内的化学药和生物制品,包括创新药、仿制药及原料药。适用于大型药企、Biotech公司以及CRO和CDMO等企业。 要点总结: - 药品配制:规定了自愿外包设施的标准和要求,以提高药品配制的质量和安全性。
- 供应链安全:建立了药品供应链的国家标准,要求增强药品分配的安全性。
- 批发分销商标准:为处方药批发分销商设定了全国性标准。
- 第三方物流提供商:规定了第三方物流提供商的国家标准和统一的国家政策。
- 违规处罚:对违反新法规定的个人和企业,明确了处罚措施。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - QA:应深入理解FDARA对药品和生物制品监管要求的更新,确保公司产品符合最新的法规标准。
- 注册:需关注FDARA对药品注册流程的影响,及时调整注册策略。
- 研发:应考虑FDARA对新药研发的指导,特别是在儿科药品和设备方面。
文件适用范围:
本文适用于美国境内的化学药、生物制品、仿制药、生物类似药和医疗器械,主要针对大型药企、Biotech公司以及CRO和CDMO等企业。 文件要点总结: - 用户费用计划修订: FDARA修订并延长了处方药、医疗器械、仿制药和生物类似产品的用户费用计划。
- 信息文件开发: 要求FDA开发包括公共报告、国会报告、沟通计划等多种信息文件。
- 儿科药品和设备: 特别强调了儿科药品和设备的开发和监管,以满足特殊群体的需求。
- 药品和设备监管改进: 包括对药品审批流程和医疗器械检查的改进措施。
- 仿制药市场准入: 提出了改善仿制药市场准入的措施,以增加竞争和降低药品成本。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读建议: - 注册:了解FDASIA对药物和医疗设备注册审批流程的影响。
- 研发:关注创新药和仿制药的用户费用要求,以及对儿科药物开发的支持。
- QA:确保产品质量和安全性符合FDASIA规定的标准。
文件适用范围:
本文适用于美国境内的创新药、医疗设备、仿制药和生物类似药的注册分类,由美国食品药品监督管理局(FDA)发布,适用于Biotech、大型药企、跨国药企等各类企业。 文件要点总结: - 用户费用授权:FDASIA授权FDA从行业收取用户费用,以资助创新药、医疗器械、仿制药和生物类似药的审查工作。
- 儿科药物开发鼓励:该法案重新授权两个鼓励儿科药物开发的项目。
- PDUFA和MDUFA的第五次和第三次授权:这是处方药用户费用法案(PDUFA)的第五次授权和医疗器械用户费用法案(MDUFA)的第三次授权。
- 审查流程的稳定性和可靠性:通过这些用户费用计划,确保了审查人员队伍的稳定和审查流程的可靠性。
- 仿制药和生物类似药的用户费用计划:新计划建立在PDUFA和MDUFA成功的基础上,为仿制药和生物类似药的审查提供资金。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - QA(质量保证):必读。需关注FDORA对GMPs、产品检验、不良事件报告等方面的新要求,确保企业质量体系符合最新法规。
- 注册(注册事务):必读。需了解FDORA对药品和生物制品注册流程的影响,包括新的申报要求和加速审批路径。
- 研发(研发部门):必读。需关注FDORA对临床试验设计、数据收集和产品开发的影响,以及对罕见疾病药物开发的支持。
- 市场(市场准入):必读。需了解FDORA对市场准入的影响,包括新的标签要求和市场独占期规定。
- 临床(临床研究):必读。需关注FDORA对临床试验多样性、远程临床试验和数据收集等方面的新要求。
适用范围:
本文适用于美国市场的药物和生物制品,包括创新药、仿制药、生物类似药、原料药等,涉及药品类型包括化学药和生物制品。适用于Biotech、大型药企、跨国药企等企业类别。 要点总结:
FDORA法案对FDA监管行业产生了显著的短期和长期影响,特别是在药物和生物制品领域。法案强调了对罕见疾病治疗的重视,要求FDA发布相关报告并制定指导草案,以提高对罕见疾病的理解和治疗选择。同时,FDORA还强调了临床试验多样性的重要性,要求申办方提交多样性行动计划,并由FDA发布相关指导。此外,法案还涉及了药品加速审批流程的现代化,包括对加速审批后的研究要求和条件的明确化,以及对未能满足这些要求的申办方的处罚措施。法案还提出了对药品和生物制品监管框架的现代化,包括对新兴技术和先进制造技术的重视,以及对临床试验设计的创新。最后,FDORA还强调了FDA在确保药品供应链安全和响应能力方面的责任,特别是在婴儿配方奶粉短缺事件后,对婴儿配方奶粉和医疗食品的监管提出了新的要求。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |