|
首页
>
资讯
>
EMA 推出欧洲临床试验互动地图
出自识林
EMA 推出欧洲临床试验互动地图
2025-03-07
3月3日,欧洲药品管理局(EMA)宣布在其欧盟临床试验信息系统(CTIS)中新增互动地图功能,为患者和医疗专业人士提供更便捷、更透明的临床试验信息获取渠道。
该地图整合了CTIS中的实时信息,以直观的仪表盘形式展示正在进行的临床试验。
CTIS作为欧盟临床试验申请的唯一入口,集成了数据库和公开门户,负责所有临床试验的提交和监管。此次新增的地图功能,是对2024年1月举办的临床试验分析研讨会中利益相关者反馈的直接回应。当时,与会者强调开发一个简单、患者友好的仪表盘的必要性,以便患者和医疗专业人士能够轻松找到在欧盟进行的临床试验。EMA还将于3月7日举办网络研讨会,实时演示地图功能,并解答利益相关者的相关问题。这一活动预计将吸引更多患者和专业人士关注并利用这一新工具。
点击“链接”,或通过识林页面可查看欧洲临床地图。
功能简约但实用,关键在于直观
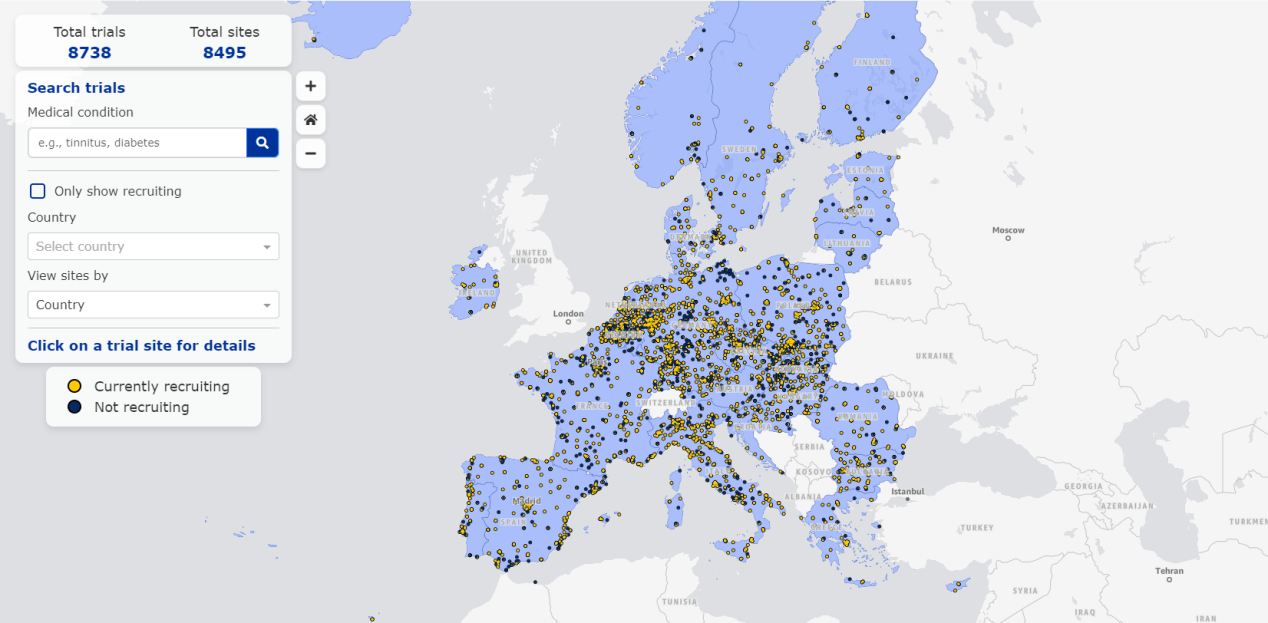
截至3月3日上线时,欧盟临床试验地图已涵盖8,738项试验,涉及8,495个试验场地。用户可以通过地图视图,查找正在招募新参与者的试验(用黄色点表示)以及招募已结束的试验(用蓝色点表示)。此外,用户还可以根据特定的欧盟/欧洲经济区国家、医学术语或常见疾病名称进行搜索。
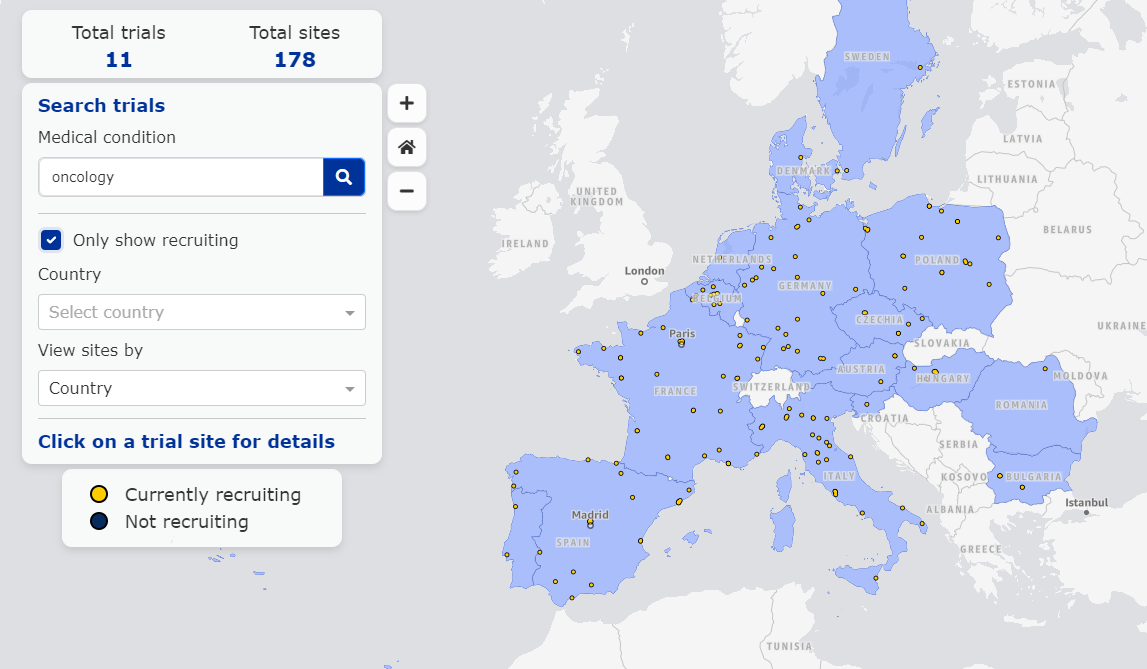
例如,搜索“Oncology”可以看到,目前欧洲有25项临床,涉及280个场地,筛选后发现其中178个场地的11项试验正在招募。
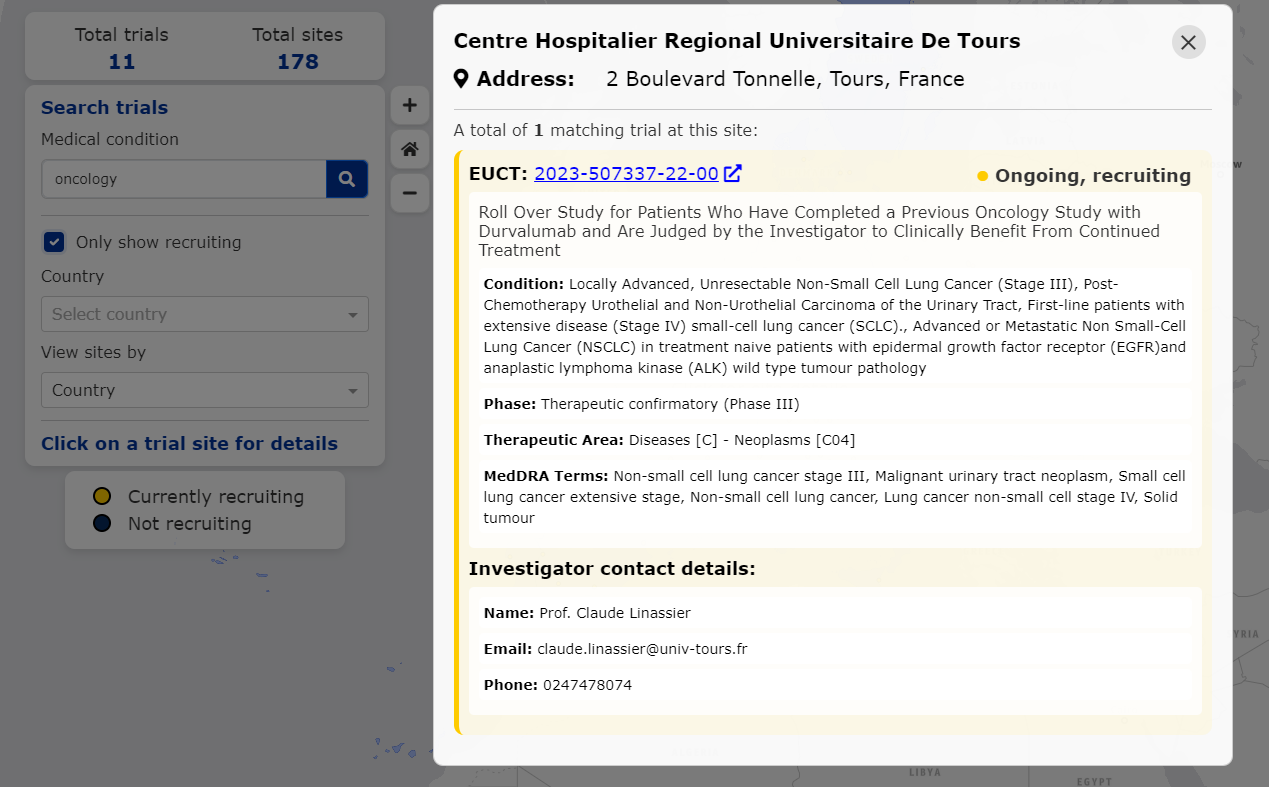
进一步点击法国地图上一个黄色标记,会弹出一个包含试验详细信息的窗口,包括试验地点地址、试验全称、疾病条件以及试验研究者的联系方式。公众可以直接通过这些信息询问有关特定试验的参与资格。用户还可以点击试验ID,直接跳转CTIS数据库获取该研究的完整信息,包括纳入和排除标准、试验终点以及赞助商在CTIS中列出的试验文件。
尽管目前欧盟临床试验地图的信息仅提供英文版本,但EMA计划在未来将其扩展至更多欧盟官方语言,以进一步提升信息的可及性和易用性。
欧盟一向关注临床试验透明度。CTIS 门户网站于 2022 年 1 月推出,是 ACT EU 计划的主要优先事项,目标是改变临床试验的启动和运行方式,协调临床试验申请的提交和评估,提高试验透明度。临床试验申办人现在必须使用该门户网站申请授权开展新的试验。
临床试验信息披露仍面临挑战
临床试验信息披露已成为全球监管的关注重点。不仅是为了满足法规的合规性,更是为了推动行业合作、加速药物研发以及提升患者对临床试验的参与度。临床试验信息披露和透明化要求最早可追溯至1997年美国《FDA现代化法案》(FDAMA)。该法案要求美国国立卫生研究院(NIH)创建一个公共信息资源,即ClinicalTrials.gov(于2000年创建),用于跟踪由FDA监管的联邦和私人资助的临床试验。随后,2007年的《FDA修正法案》(FDAAA)进一步扩展了ClinicalTrials.gov的要求,规定试验赞助商必须在试验结束后的12个月内公布结果、协议和统计分析计划。
2013年,美国制药研究与制造商协会(PhRMA)和欧洲制药工业协会联合会(EFPIA)联合发布了“负责任的临床试验数据共享原则”,强调在保护患者隐私、尊重国家监管体系完整性的同时,增强数据共享。2020年,FDA发布了关于ClinicalTrials.gov的民事罚款指导意见,规定未能按时注册和公布临床试验结果的赞助商将面临最高每天1万美元的罚款。
尽管法规不断完善,但临床试验数据的披露仍面临诸多挑战。根据2021年的一项分析,2016-2017 两年间,只有 26% 的制药商公开了所有用于获得药物批准的研究结果。67% 的药物在获得 FDA 批准后 6 个月内公布了临床试验结果,但只有 58% 符合FDAAA法案的临床试验披露要求。11% 的药物在批准时没有公布任何法律要求报告结果的试验。整体来说,大企业比小企业更加透明。在所有试验结果的公开可用方面,生物制品和药品之间存在统计学上的显著差异:85% 对 47%。值得注意的是,分析报告中几乎所有的生物制品(22 个中的 19 个)都是由市值较高的大企业开发的。
识林-实木
识林®版权所有,未经许可不得转载
法规指南解读 适用岗位: - 必读岗位:注册专员(Regulatory Affairs Specialist)、质量保证专员(QA)、研发人员(R&D)、临床研究协调员(CRC)。
- 工作建议:
- 注册专员:熟悉FDA现代化法案的条款,确保注册流程符合最新法规要求。
- 质量保证专员:监控法规变化,更新质量管理体系。
- 研发人员:在药物开发过程中考虑法规要求,确保研发合规。
- 临床研究协调员:确保临床试验遵循FDA现代化法案规定。
适用范围:
本文适用于美国药品、生物制品、医疗器械和食品的监管,适用于所有在美国运营的药企、生物科技公司、CRO和CDMO等。 文件要点总结: - 法规修订:明确了对《联邦食品、药品和化妆品法》及《公共卫生服务法》的修订,以改善食品、药品、医疗器械和生物制品的监管。
- 定义明确:为“药品”、“医疗器械”、“食品”和“膳食补充剂”等术语提供了明确的定义。
- 监管改善:分为五个部分,分别针对药品、医疗器械、食品的监管进行改善,并包含一般规定和生效日期。
- 强调合规:通过修订法规,强调了对药品、医疗器械和食品的合规性要求。
- 监管现代化:旨在通过现代化法案,提高监管效率和确保公共健康安全。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位建议: - RA(注册):了解FDA对处方药和医疗器械用户费项目的修订和延期,以及对药品上市后安全监管的增强措施。
- QA(质量管理):关注FDA对药品上市后安全性的监管增强措施,确保企业合规。
- 研发:了解儿科药品研究和医疗设备安全改进的相关要求,指导研发工作。
适用范围:
本文适用于美国境内的化学药、生物制品、疫苗、中药等药品类型,涉及创新药、仿制药、生物类似药、原料药等注册分类。适用于Biotech、大型药企、跨国药企、CRO和CDMO等各类企业。 文件要点总结: - 用户费项目修订:明确了对处方药和医疗器械用户费项目的修订和延期,以支持FDA的监管工作。
- 儿科药品研究:特别强调了儿科药品研究的重要性,通过《儿科医疗设备安全和改进法案》和《儿科研究公平法案》来增强儿科药品的安全性和有效性。
- 上市后药品安全监管:通过《增强药品上市后安全监管法案》增强了FDA对药品上市后安全性的监管权力。
- 临床试验数据库:规定了临床试验数据库的建立和维护,以提高临床试验的透明度和可访问性。
- 食品和药品安全:涵盖了食品和药品安全的相关条款,以保护公众健康。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |