首页
>
资讯
>
2015 - 2024年欧美日等六家监管机构新药批准趋势简报
出自识林
2015 - 2024年欧美日等六家监管机构新药批准趋势简报
2025-08-12
近日,国际组织监管科学创新中心(Centre for Innovation in Regulatory Science, CIRS)发布了其年度分析报告 ,聚焦于2015至2024年间EMA、FDA、日本药品和医疗器械管理局(Pharmaceuticals and Medical Devices Agency, PMDA)、加拿大卫生部(Health Canada,HC)、瑞士医药管理局(Swissmedic)以及澳大利亚治疗产品管理局(Therapeutic Goods Administration, TGA)六大监管机构对新活性物质(New Active Substance, NAS)的批准情况。
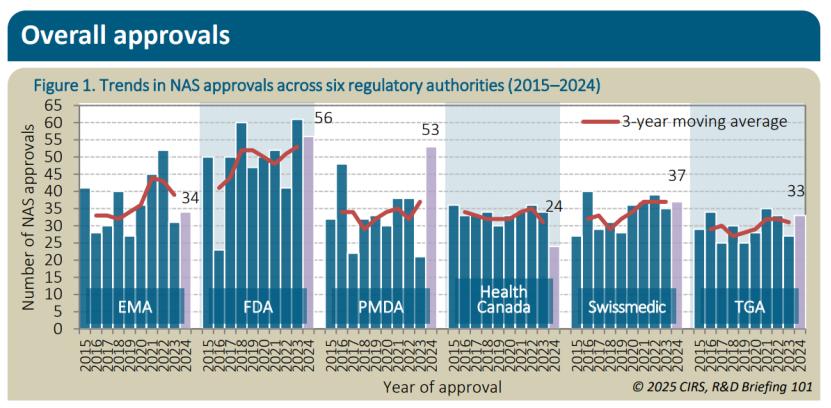
2024年FDA批准的NAS数量最多,达到56个,其次是PMDA(53个)、Swissmedic(37个)、EMA(34个)、TGA(33个)和HC(24个)。在过去十年中,FDA批准的产品数量超过其他机构,但并非所有药品都能迅速实现国际化。在提交时间间隔方面,2024年FDA的中位提交间隔时间最短,为0天,表明至少有一半的NAS首先提交至FDA,其次是EMA(49天)、TGA(219天)、HC(262天)、Swissmedic(417天)和PMDA(727天)。援引Nature子刊文章《2015–2024中国制药行业崛起:十年创新》 ,我国在2024年批准了93个创新药(与本报告NAS定义不完全一致),其中42%是国内研发 。
FDA相对较快,走加速路径也最多
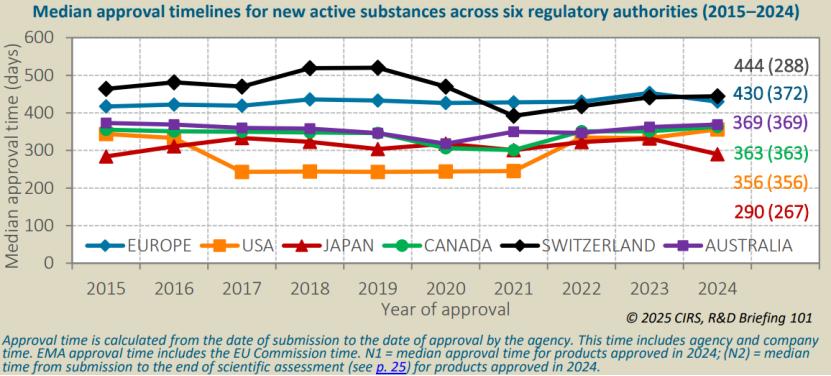
尽管过去20年各机构均有所进步,但六大监管机构的NAS中位批准时间仍存差异。2024年PMDA的中位批准时间最短,为290天,随后依次是FDA(356天)、HC(363天)、TGA(369天)、EMA(430天)和Swissmedic(444天)。PMDA与Swissmedic之间的中位批准时间最大差异达154天。但看多年数据,还是FDA批得相对较快。
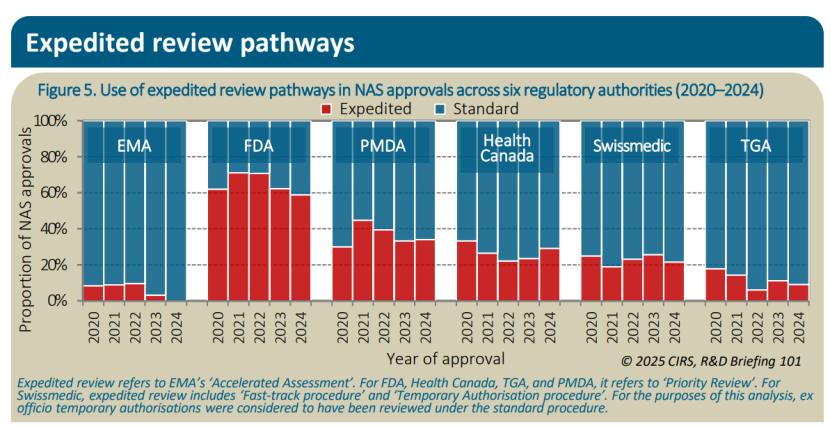
报告还显示,2024年各机构在NAS批准中使用加速审评 路径的比例各不相同。FDA的使用比例最高,达到59%,其次是PMDA(34%)、HC(29%)、Swissmedic(22%)和TGA(9%)。EMA因申请撤回或回归标准审评,未通过加速审评批准任何NAS。2024年,EMA和FDA通过附条件、临时、暂定路径批准的NAS比例均为18%,Swissmedic为14%,TGA为12%,HC为8%。
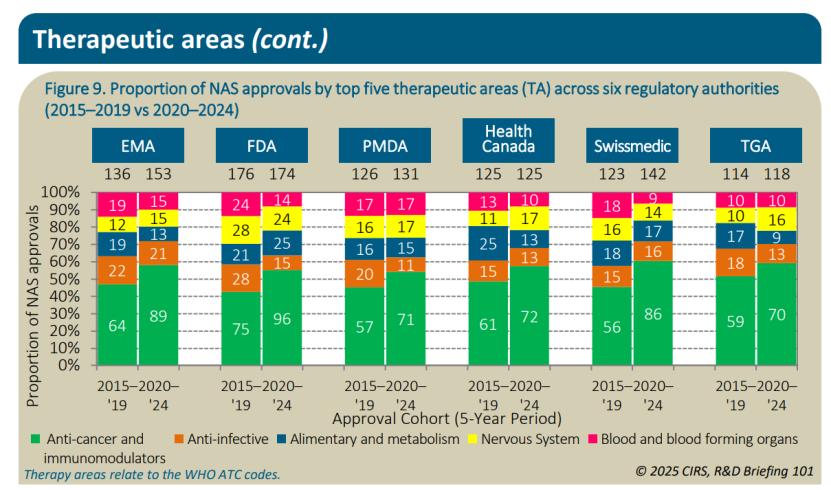
从TOP 5治疗领域看,2020至2024年,抗感染疗法在六大监管机构中的中位批准时间最短,为284天,其次是消化和代谢疗法(310天)、抗癌和免疫调节疗法(349天)、血液和造血器官(362天)以及神经系统(380天)。在过去十年中,抗癌和免疫调节疗法在六大监管机构的NAS批准中占比最高,从2015至2019年的47%增长至2020至2024年的57%,而其他四个领域则保持稳定或有所下降。
再聚焦FDA,看其审评发补的细节
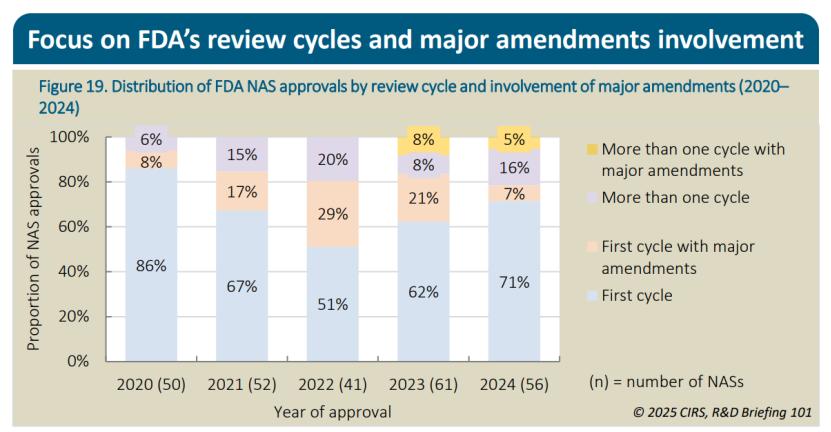
尽管在2022年,FDA首次审批周期内批准NAS的比例降至51%的低点,但此后呈现出稳步回升态势,2023年上升至62%,2024年进一步增至71%。2022年,重大增补 (大概相当于我国的“发补”)的使用频率高于其他任何一年,这有助于申办方尽早修正缺陷 ,避免收到“完全回应函”(CRL )。这种做法帮助FDA将NAS在首次审评周期内完成批准的比例保持在约80%。2020至2022年期间,没有涉及重大增补的NAS在多次审评周期后获批(换句话说,即使有多次发补机会也未能最终说服FDA),但这种情况在2023年和2024年发生了变化,分别有8%和5%在多次审评周期后获批。
援引《2025年 RDPAC 药品注册时限调研报告》 ,2024年我国进口药品的数据是,生物制品 首次发补率为95.8%,化药首次递交的创新药以及非2.4类改良型新药 100%发补,5.1类发补率62.5%。
除了上述审评路径相关数据,CIRS报告还提供了一个特别的角度,即从患者观点/患者体验数据(patient experience data,PED)上审视FDA批准的NAS。
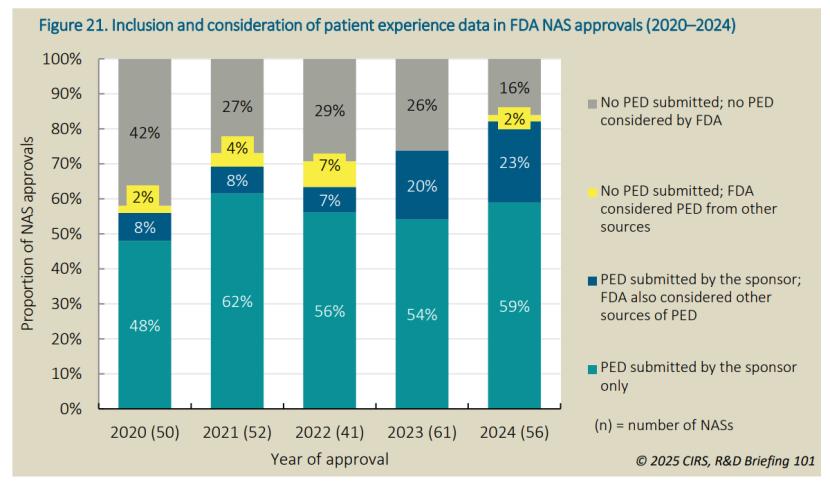
近年来,FDA将PED作为“以患者为中心的药物研发” (PFDD )计划的一部分,越来越多地将其纳入监管决策。2020至2024年,纳入和/或考虑PED的NAS比例从58%稳步上升至84%,这一增长主要来自“申办方提交PED并且FDA也识别其他来源的PED”,从2020年的8%上升至2024年的23%。仅基于申办方提交的PED的批准比例也从2020年的48%上升至2024年的59%。相比之下,仅由FDA独立考虑外部PED(申办方未在申请中提交)的情况在2020至2024年间波动,2024年仅占2%。简言之,PED主要还是靠申办方主动提交。
不过,报告指出,由于缺乏关于PED在监管审评中如何被权衡的公开细节,很难评估其对审评流程的实际影响。因此,数据仅供参考。
更多细节内容,可参见报告原文。
识林-实木
识林® 版权所有,未经许可不得转载