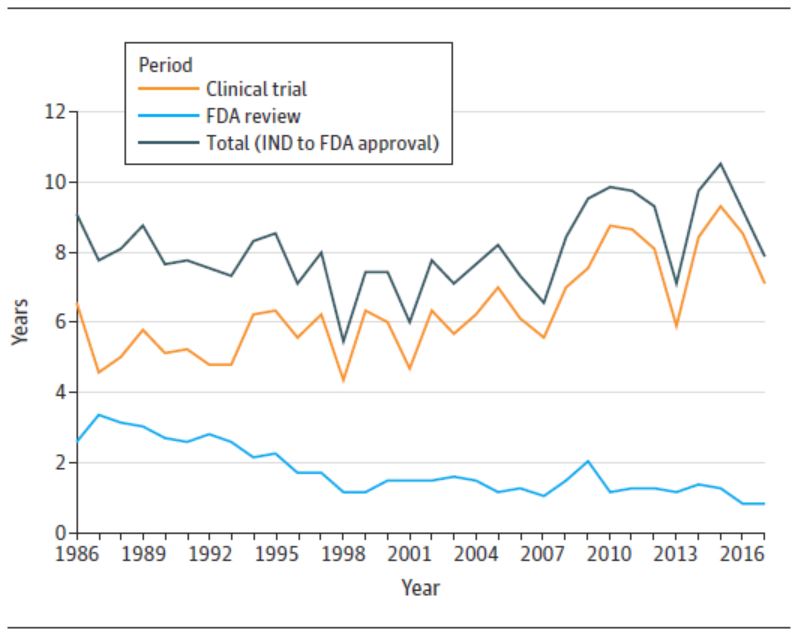
不过有一类产品的开发时间逆转了这一趋势,获得突破性治疗认定的药比其它药短得多,到 2016 年这类药物从开发到获批的时间不到五年。突破性治疗药物开发时间短可能是由于 FDA 根据较少且较早阶段的研究批准了这些产品。作者引用了以前的研究指出,“一项研究发现,2013-2016 年间批准的所有突破性治疗认定药物中,有 52%(16/31)是根据 I 期或 II 期临床试验批准的,而 45%(14/31)是基于一项试验批准的,42%(13/31)没有使用活性或安慰剂对照。”作者指出,这些结果对于肿瘤药更为明显。
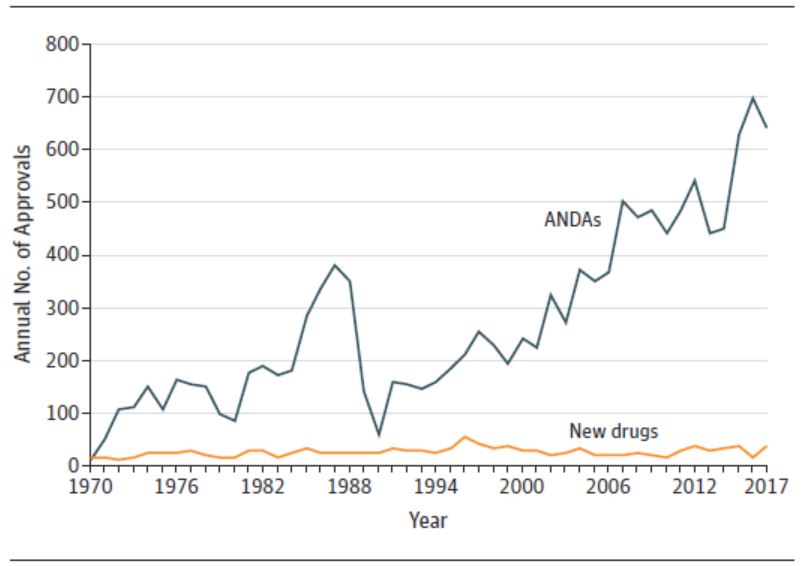
另外,研究发现 FDA 批准的药物中孤儿药所占比例也大幅增加,从 1984-1995 年的 18% 到 2008-2018 年的 41%。作者指出,对许多疾病遗传基础理解的进步将越来越有可能将常见疾病(例如,肺癌)分为较小的、遗传定义的亚型。
他表示,“如果 FDA 过去四十年改革的重点是更快地找到新治疗药物,那么对 FDA 这些改革开展再次改革的重点应该是提高这些计划的有效性和效率。”Sharfstein 表示,总体而言,文章中的数据并未显示出 FDA 正陷于困境,只是随着时间的推移监管程序已经演变成一系列特殊计划、灵活的审评标准和慷慨的激励措施。
Sharfstein 在评论中提出了四点建议:
首先,“国会和 FDA 应该对各种加快审评计划进行合理化改革,”以解决围绕孤儿药的竞争问题,并确保获得快速通道和突破性认定的产品更有可能为患者带来更大获益。
参考资料
[1] Darrow JJ, Avorn J, Kesselheim AS. FDA Approval and Regulation of Pharmaceuticals, 1983-2018. JAMA. 2020;323(2):164–176. doi:10.1001/jama.2019.20288
[2] Sharfstein JM. Reform at the FDA—In Need of Reform. JAMA. 2020;323(2):123–124. doi:10.1001/jama.2019.20538