|
首页
>
资讯
>
英国 MHRA 2018 GMP 检查缺陷趋势分析:数据总览
出自识林
英国 MHRA 2018 GMP 检查缺陷趋势分析:数据总览
2020-05-26
英国药品与医疗保健产品监管机构(MHRA)于 2019 年 10 月发布了其在 2018 年的 GMP 检查缺陷,与前几年不同,2018 年 GMP 检查缺陷数据采用了电子表格的形式,公布了 6200 多行数据,人们可以根据自己的需要进行解析和呈现数据。而此前的 2015 年 和 2016 年,MHRA 均是提供的约 100 张带有表格、图片和文字的幻灯片报告,2017 年未发布任何数据。 和 2016 年,MHRA 均是提供的约 100 张带有表格、图片和文字的幻灯片报告,2017 年未发布任何数据。
本文对 MHRA 公布的缺陷数据以图表的方式分析呈现出一些趋势,希望帮助企业作为自查和持续改进的一部分,对照缺陷发现开展自评,提前发现问题并改进和完善。
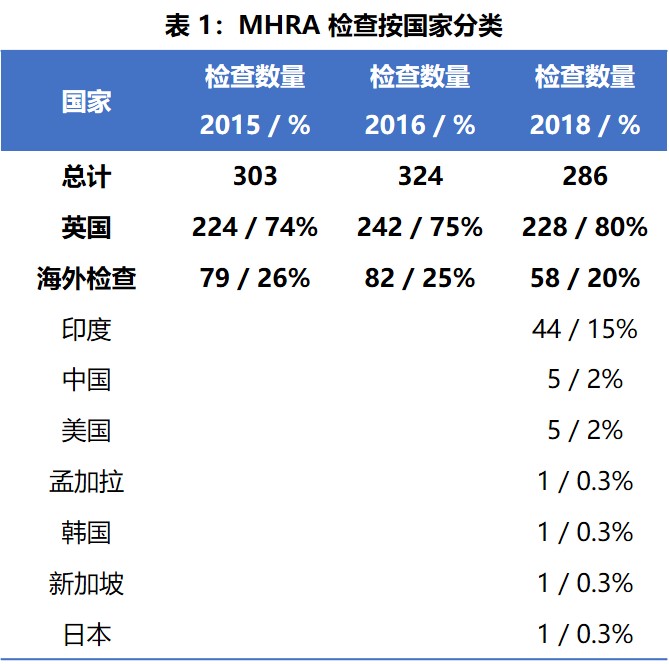
MHRA 在 2018 年对英国国内和国外执行了 286 次 GMP 检查。英国以外的检查在中国、印度、美国、日本、新加坡、韩国以及孟加拉国进行。2015 年和 2016 年 MHRA 报告未提供针对英国和“海外”特定国家/地区的数据。与 2015 年和 2016 年相比,2018 年的检查次数有所减少。
检查分布
表1 列出了 MHRA 在 2018 年间按国家分类的药品检查数量。与往年一样,几乎大部分检查都在英国国内。2018 年在英国执行的检查百分比与 2015 年和 2016 年相比略有增加,海外检查的比例在 2018 年略有下降(20%)。2018 年,在英国以外进行的检查数量最多的是印度,总共进行了 44 次检查。其余 14 次境外检查在六个国家进行,中国和美国分别有五次检查,其他国家各一次。
GMP 章节和附录
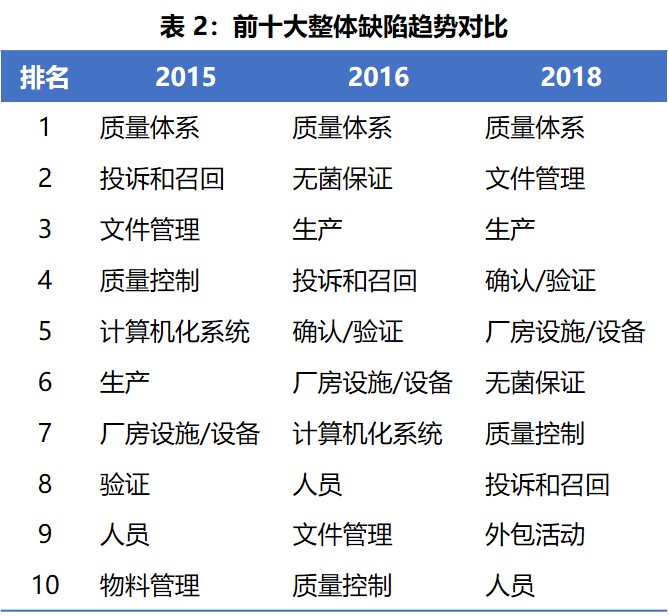
表 2 显示了 2015、2016 和 2018 所有缺陷中出现最多的欧盟 GMP 章节和附录趋势对比。质量体系在三年中均位居榜首。与前两年相比,2018 年的显著变化包括:
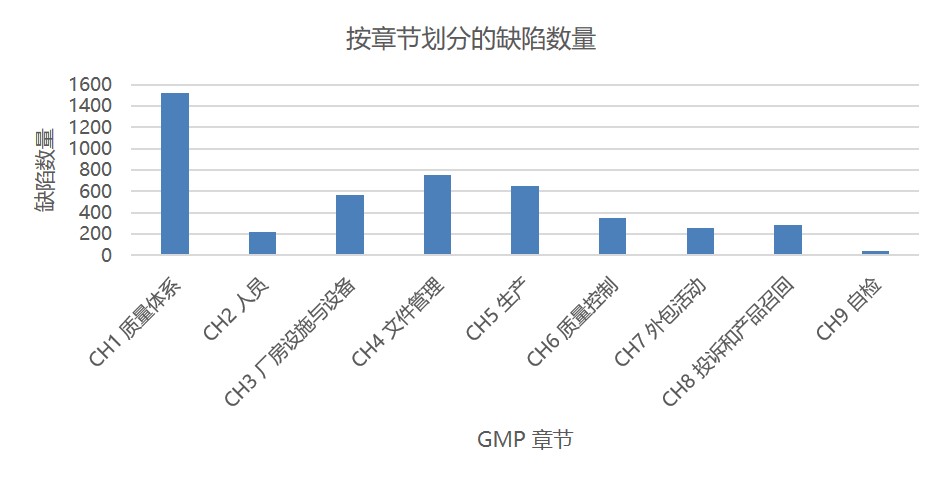
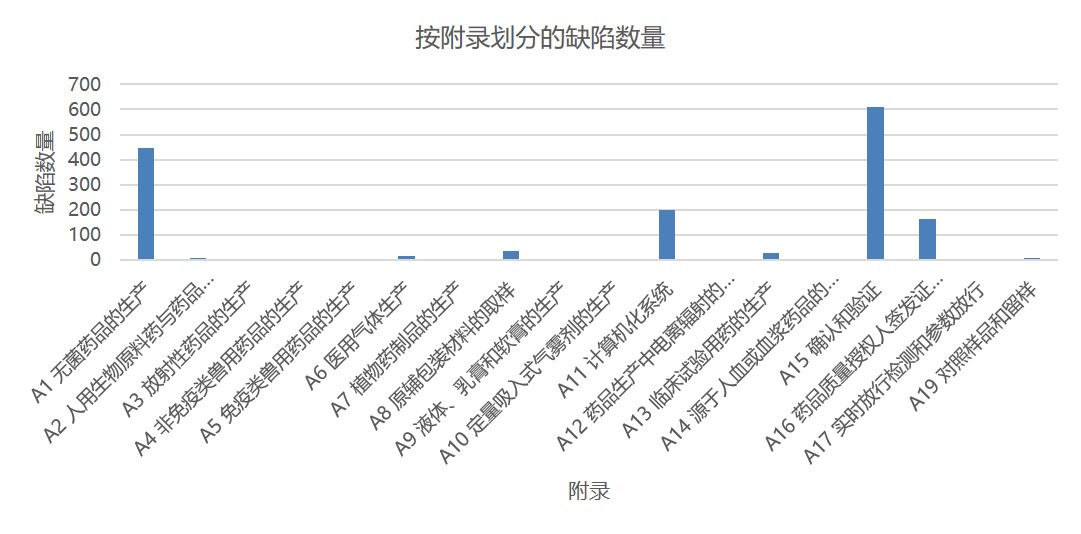
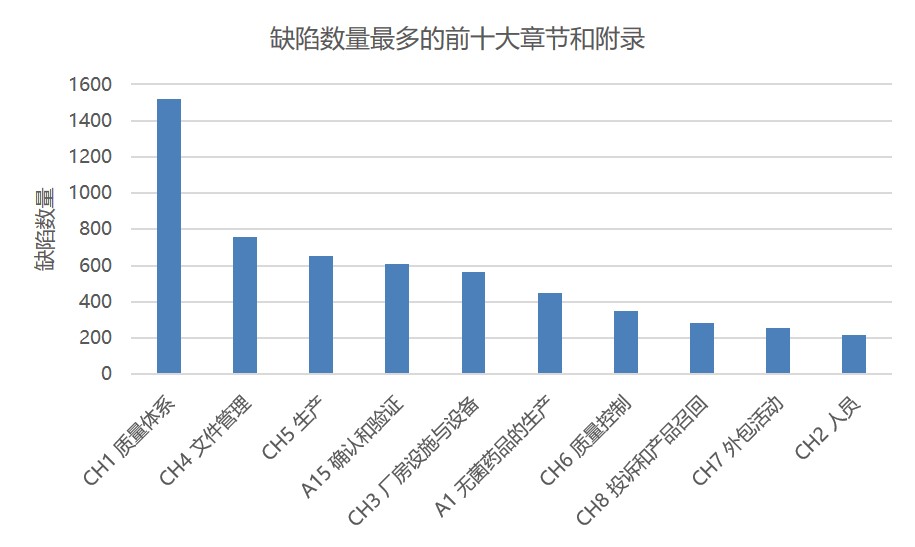
图 1 和图 2 分别显示了根据所引欧盟 GMP 章节或附录的缺陷总数。这些数据包括所有缺陷分类:严重、主要和其它。附录 15 确认和验证以及附录 1 无菌保证分别位居第一和第二位,分别包括大约 600 条和 450 条缺陷。其次是附录 11 计算机化系统和附录 16 药品质量授权人签发证书和放行产品。
GMP 第一章质量体系被引证约 1500 次,其缺陷引证次数是处于第二位章节的两倍多。其次是第四章文件管理有约 760 条缺陷项。然后是第五章生产 650 条缺陷项。
图 3 显示了将 GMP 附录和章节合并在一起时位居前十的类别。
严重和主要缺陷
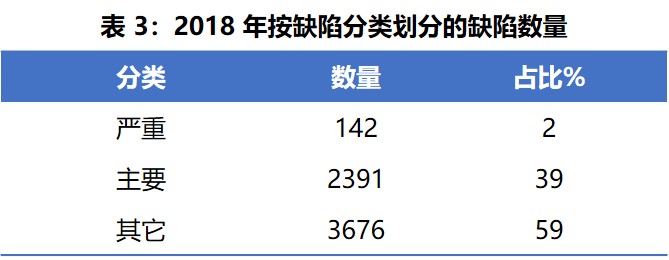
表 3 列出了 2018 年按缺陷分类的情况。严重缺陷占总数的 2%,主要缺陷占 39%,其它缺陷占 59%。
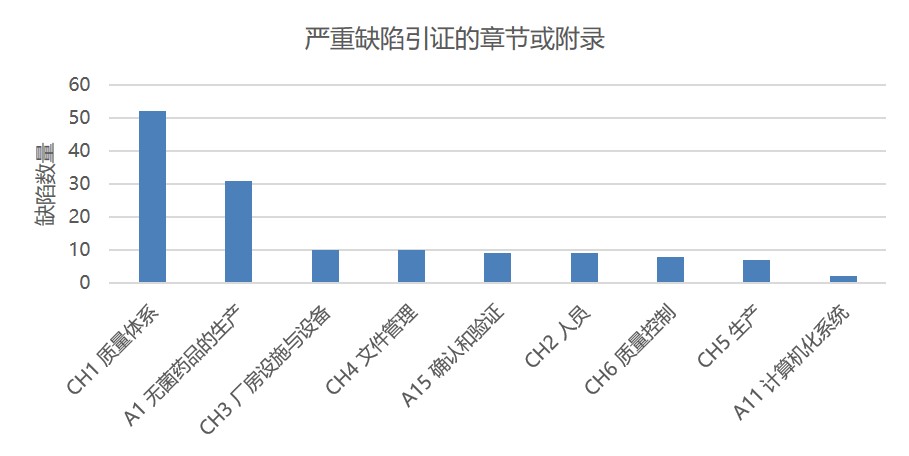
在 6200 多条缺陷中,大多数严重缺陷和主要缺陷都集中在几个章节和附录中。图4 显示了严重缺陷数量及所在章节或附录。在严重缺陷中,37% 与第一章质量体系有关,22% 与附录1无菌保证有关。
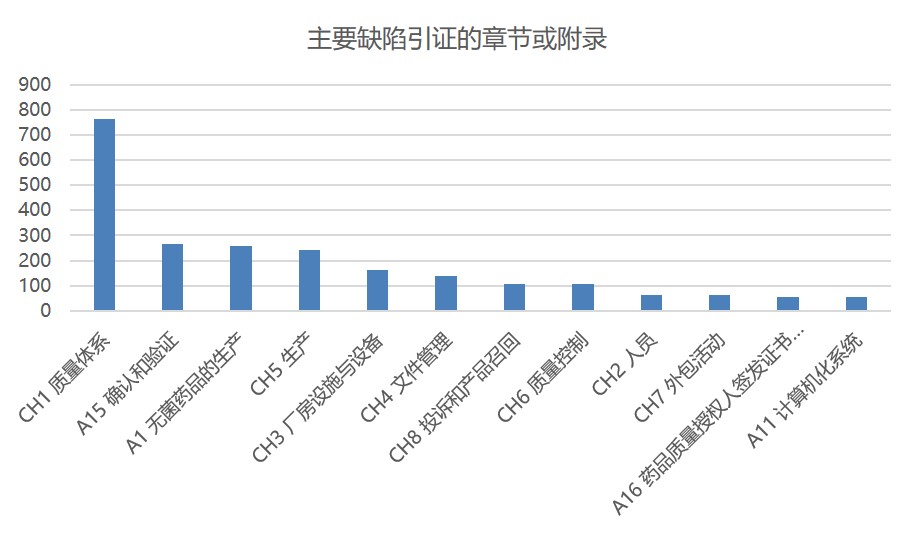
图 5 展示了与主要缺陷相关的 12 个章节和附件以及各自引证的缺陷数量。无论是所有缺陷,还是严重缺陷或者主要缺陷,质量体系章节都稳居第一,表明 MHRA 重点关注完善的质量体系对 GMP 合规性的重要性。
希望 MHRA 将会发布有关 2019 年的类似数据,我们可以将这些数据放在一起比较。
作者:识林-椒
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
适用岗位必读建议: - QA(质量保证):应深入理解附录15的要求,确保质量管理体系与GMP标准一致。
- 生产:必须遵循验证主计划(VMP)和相关文件,确保生产过程中的设备和系统符合GMP要求。
- 工程:在设计和安装阶段应确保设备、设施满足URS,并参与DQ、IQ、OQ和PQ等确认活动。
- 研发:在产品开发阶段应考虑工艺验证的要求,确保研发到生产的顺利过渡。
- 验证:负责制定和执行确认与验证计划,确保所有相关活动符合附录15的指导。
文件适用范围:
本文适用于化学药、生物制品、疫苗和中药等药品类型,包括创新药、仿制药、生物类似药和原料药等注册分类。适用于在欧盟成员国注册和生产的药物,针对Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 文件要点总结: - 确认与验证原则:强调了确认和验证活动应贯穿药品生命周期,基于质量风险管理。
- 文件记录重要性:确认和验证过程中产生的所有文件应得到适当批准,并明确文件间的相互关系。
- 设备和系统确认:从URS到DQ、IQ、OQ和PQ,每个阶段都有其特定要求和标准。
- 工艺验证方法:介绍了传统验证、同步验证、持续工艺确认和混合方法,并强调了工艺验证的科学性和风险基础。
- 变更控制:变更应通过质量风险管理评估,确保对产品质量的影响得到控制。
以上仅为部分要点,请阅读原文,深入理解监管要求。 岗位必读指南: - 质量受权人(QP):必读。确保对药品生产和放行的全过程有深入理解,并按照附录要求执行认证和放行。
- 生产管理(PM):必读。了解QP的放行要求,确保生产过程符合GMP和MA规定。
- 质量保证(QA):必读。监督和确保QP放行流程的合规性,提供必要的GMP支持。
- 注册部门:必读。理解放行要求,确保注册文件符合规定。
- 研发(R&D):了解。知晓放行流程,确保研发阶段的产品符合放行标准。
工作建议: - QP应持续接受相关产品类型、生产工艺的培训,确保认证工作的准确性和合规性。
- PM需与QP紧密合作,确保生产记录的完整性和可追溯性。
- QA应建立和维护放行流程的审计跟踪,确保所有放行活动符合GMP要求。
- 注册部门应确保所有上市许可文件与放行流程保持一致,及时更新注册信息。
文件适用范围:
本文适用于欧盟内持有上市许可或供出口的人用药品和兽用药品,包括临床试验用药(IMP)。适用于化学药、生物制品等,不包括官方控制机构批放行的血液和免疫产品。发布机构为欧盟,适用于Biotech、大型药企、跨国药企等。 要点总结: - QP认证责任:QP负责确保每批药品的生产和检查符合GMP和MA要求,包括对生产全过程的了解和认证。
- 生产和放行流程:详细规定了认证程序、第三方GMP评估、非预期偏差处理以及批放行的具体要求。
- 多场地生产协调:若生产过程涉及多个场地,QP需确保所有场地的生产活动符合GMP和MA要求,并在认证中考虑其他QP的确认。
- 进口药品的特殊要求:对于在欧盟以外生产的药品,QP需确保满足进口和认证的相关条件,包括活性成分的分析和检验。
- 记录和文档管理:QP认证的记录必须保持更新,并在规定的时间内可供监管机构检查。
以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读 适用岗位及工作建议: - QA(质量保证):必读。需确保所有生产操作符合GMP原则,并监督执行。
- 生产:必读。应按照明确的规程进行生产,并确保产品质量。
- 注册:必读。了解生产限制对市场供应的影响,确保及时通知相关方。
- 研发:参考。了解生产过程中的质量管理要求,以指导产品研发。
适用范围:
本文适用于化学药品、生物制品、原料药等的生产,特别强调了防止交叉污染的措施和供应商确认的重要性。适用于欧盟地区内所有药品生产企业,包括Biotech、大型药企、跨国药企等。 文件要点总结: - 生产操作规程:生产操作必须遵守明确的规程,符合GMP原则,确保产品质量符合生产和市场授权要求。
- 防止交叉污染:新增了关于预防交叉污染的详细措施,强调了质量风险管理在控制交叉污染风险中的应用。
- 供应商确认:新增了供应商确认的要求,以反映药品生产许可持有人确保原料药生产符合GMP的法律责任。
- 药品短缺通知:新增了供应受限制时应通知官方的内容,以应对药品短缺问题。
- 验证和质量控制:强调了关键工艺和规程应定期进行再验证,确保其仍可达到预定结果,并要求所有物料和产品在放行使用或放行发运前必须经过质量控制部门的检查和放行。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - QA(质量保证部门):负责解读和实施GMP指南,确保投诉和召回流程符合规定。
- QC(质量控制部门):参与质量缺陷的调查和评估,确保产品质量。
- QAP(质量授权人):在必要时参与投诉和召回的决策过程。
- 注册部门:了解法规要求,确保产品注册信息的合规性。
- 研发部门:在产品开发阶段考虑质量风险管理。
工作建议: - QA:制定和更新SOP,确保所有投诉和召回活动符合GMP要求。
- QC:对投诉中涉及的产品进行彻底的质量检查和风险评估。
- QAP:在召回决策中提供专业意见,确保产品质量和患者安全。
- 注册部门:确保所有产品注册信息与GMP要求一致,及时更新注册资料。
- 研发部门:在产品设计中考虑质量风险,减少潜在的投诉和召回风险。
适用范围:
本文适用于欧盟境内的化学药和生物制品,包括创新药、仿制药、生物类似药及原料药。适用于Biotech、大型药企、跨国药企以及CRO和CDMO等各类企业。 要点总结:
新版EU GMP第八章强调了在处理投诉、质量缺陷和产品召回时应用质量风险管理原则。要求建立系统和程序记录、评估、调查和审查投诉,必要时迅速召回人用或兽用药品。明确了向监管机构报告质量缺陷的责任,并在外包活动中明确各方角色和责任。规定了投诉和质量缺陷处理的人员和组织要求,强调了跨学科团队的重要性。详细说明了投诉和质量缺陷调查的操作规程,包括描述、范围确定、风险评估、决策过程、沟通和根本原因分析。强调了在召回决策中考虑风险评估和风险转移,保证关键药品供应。要求建立详细的书面程序,包括质量缺陷描述、范围确定、样品处理、风险评估、决策和沟通、根本原因分析和CAPA。强调了模拟召回的重要性,并要求对所有可能的原因进行彻底调查,以制定有效的预防措施。 以上仅为部分要点,请阅读原文,深入理解监管要求。 |