|
首页
>
资讯
>
细胞与基因治疗产品常见 GMP 缺陷及质量管理挑战
出自识林
细胞与基因治疗产品常见 GMP 缺陷及质量管理挑战
2020-06-12
美国 FDA 检查员表示在对细胞和基因治疗产品的检查中更常发现设施和生产缺陷,而行业则表示,药品 GMP 并不总是适用于细胞和基因产品。
6 月 1 日举行的 ISPE 生物药制造网络研讨会上,FDA 官员在研究中指出,在 483 表报告的 GMP 缺陷中,比起其它类型的产品,细胞和基因治疗产品制造商的设施和生产方面的缺陷更多。而随后演讲的行业代表则认为细胞和基因治疗产品在遵守药品 GMP 要求方面的一些挑战,药品 GMP 要求更多的是为小分子和大分子制药商设计的,而对于细胞和基因产品则存在一定的限制。
FDA 目前并没有专门针对细胞和基因治疗产品的 GMP 法规或指南,这类产品的制药商应遵循以下法律法规要求:
- 《联邦食品、药品和化妆品法案》第501(a)(2)(b)条以及 21 CFR 210 和 211 中药品相关的 GMP 规定;
- 21 CFR 600 有关生物制品的法规;
- 21 CFR 1271 人类细胞、组织以及基于细胞组织的产品法规
欧盟于 2017 年 11 月颁布了关于先进治疗医药产品(ATMP)的 GMP 定稿指南并于 2018 年 5 月实施。【欧盟委员会最终发布关于先进治疗医药产品的 GMP 标准 2017/11/26】
国际药品认证合作组织(PIC/S)则于去年 10 月现有 GMP 附录 2,其中附录 2A 涵盖 ATMP 产品,并向公众咨询意见。【PICS 拟拆分 GMP 附录 2 以包括先进治疗产品 2019/10/07】罗氏欧洲、中东和非洲以及日本对外关系主管 Lina Ertle 在会上介绍了欧盟 ATMP 定稿指南与 PIC/S 附录 2A 征求意见稿的异同,并介绍了 ISPE 对 PIC/S 附录 2A 的反馈意见。
我国药品审核查验中心于去年 11 月发布《GMP 附录-细胞治疗产品》征求意见稿。
GMP 缺陷
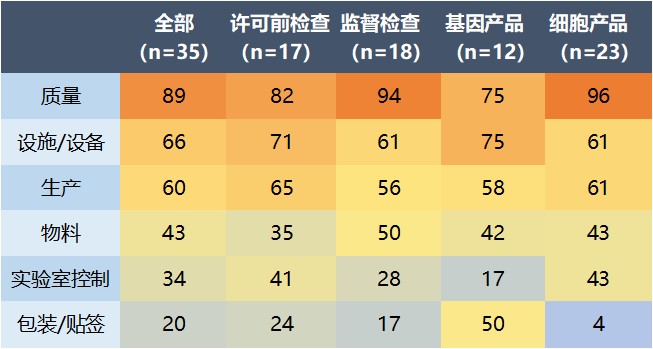
FDA 生物制品审评与研究中心(CBER)监管项目经理和 CMC 设施审评员 Ekaterina Allen 在演讲中总结了 2007-2020 年间对细胞和基因治疗产品制造商检查发布的 483 表中的一些主要缺陷。总共对 17 所设施执行了 35 次检查,包括许可前检查和监督检查。其中大多数是美国国内检查。
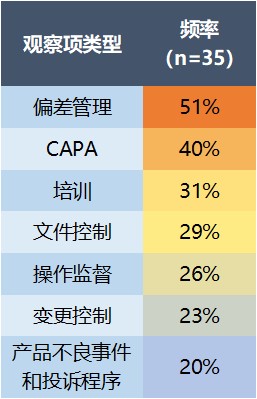
数据显示,所有针对细胞和基因治疗产品制造商的检查中,第一大缺陷类别是质量系统缺陷(占 89%),其中 51% 为偏差管理问题,主要是没有启动偏差调查。在质量系统缺陷中排在第二位的则是纠正预防措施(CAPA)不到位(40%),然后是对员工的培训不足(31%)。
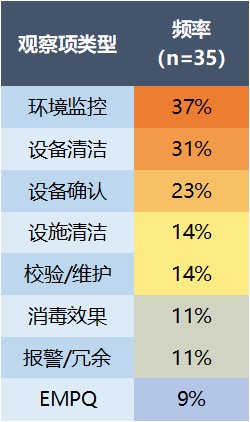
第二大缺陷类别是设施/设备问题(66%)。在这一类别中,37% 集中在环境监控不足上,31% 是设备清洁问题,23% 是设备确认问题。
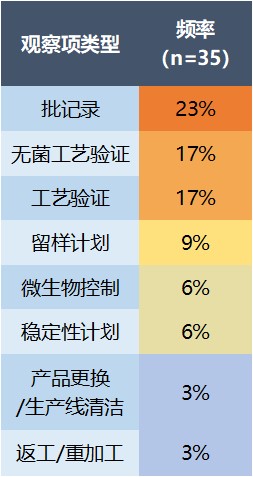
第三大缺陷类别是生产问题(60%)。在生产系统中发现的问题中,有 23% 属于批记录不足,17% 为无菌工艺验证缺失或不充分。
为避免这些 GMP 问题,Allen 建议生产商按照 GMP 的要求“记录你所做的事情,做你所记录的事情。”Allen 指出,细胞和基因治疗产品制造商在设施和生产方面的一些缺陷更为独有,而质量体系方面的缺陷则更具共性,其它产品制造商也存在相似的问题。
细胞和基因治疗产品在满足 GMP 方面的挑战
诺华公司的细胞和基因技术开发与生产质量战略规划主管 Luciana Mansolelli 则在其演讲中表示,在细胞和基因治疗产品领域满足 GMP 要求并不总是那么容易。对这些产品“使用传统 GMP 方法可能并不总是有效的。”例如,传统产品的放行检测没有考虑到这些产品必须在较短的周转时间内向患者提供。她表示,“可能需要特殊的放行路径。”
Mansolelli 还敦促行业利用与 FDA 的早期会议尽早解决所有问题。这些早期会议包括初始有针对性的监管建议参与(INTERACT)和 CBER 的先进技术团队(CATT)计划。可能讨论的领域包括安全性问题以及化学、生产和控制(CMC)所需的数据集。她分享表示,对于诺华而言,与监管机构尽早举行会议对于他们的药品注册至关重要。
作者:识林-椒
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
适用岗位: - QA(质量保证部门):必读。负责确保所有生产活动符合GMP要求,并监督质量体系的实施。
- 生产(Production):必读。需要遵循指南中的GMP要求,特别是在人员、环境和设备方面的规定。
- 研发(R&D):必读。在开发ATMPs时,必须遵守指南中的GMP要求,尤其是在风险评估和产品质量方面。
- 注册(Registration):必读。需要了解GMP指南,以确保注册文件和市场授权产品符合GMP标准。
- 质量控制(QC):必读。负责执行质量控制测试,并确保所有测试符合GMP要求。
工作建议: - QA:制定和更新内部SOPs以符合GMP指南,监控生产过程以确保合规性,并提供员工培训。
- 生产:确保生产环境、设备和人员符合GMP要求,特别是在无菌操作和交叉污染控制方面。
- 研发:在产品开发阶段就考虑GMP要求,特别是在原材料选择和生产工艺设计方面。
- 注册:确保所有注册文件和市场授权产品符合GMP指南,特别是在产品规格和生产过程描述方面。
- 质量控制:根据GMP指南制定和执行质量控制测试,确保所有测试结果准确记录并符合规定标准。
适用范围:
本文适用于欧洲经济区内的先进治疗药物产品(ATMPs),包括基因治疗产品、体细胞治疗产品和组织工程产品。适用于已获得市场授权的ATMPs和用于临床试验的ATMPs。适用于Biotech、大型药企、跨国药企等企业类别。 要点总结: - GMP合规性:强调所有获得市场授权的ATMPs必须遵守GMP,临床试验用ATMPs也必须符合GMP。
- 风险评估方法:明确ATMP制造商需应用基于风险的方法,以确保产品质量、安全和效果。
- 人员和培训:强调人员培训的重要性,包括GMP原则和特定于ATMPs的生产、测试和可追溯性要求。
- 环境和设施:详细规定了生产区域的设计和建设要求,包括无菌环境和环境监控。
- 文档和记录:要求建立充分的文档系统,确保材料、中间产品和最终产品的适当规范,并保持适当的记录。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |