|
首页
>
资讯
>
日本 PMDA 发布2024年 GMP 年报,检查中国最多,分析关键缺陷
出自识林
日本 PMDA 发布2024年 GMP 年报,检查中国最多,分析关键缺陷
2025-07-11
7月7日,日本药品和医疗器械管理局(PMDA)发布《2024年GMP/GCTP年度报告》。该报告详细总结了PMDA在过去一年中对药品生产质量管理规范(GMP)及基因、细胞和组织产品生产质量管理规范(GCTP)的监管工作成果。报告还随附了一份典型缺陷清单,共计43项缺陷。
目前报告是日文版,PMDA称未来会有英文翻译,但2023年报英文版曾延后5个月才发布。
大部分是书面检查,对中国实地检查最多
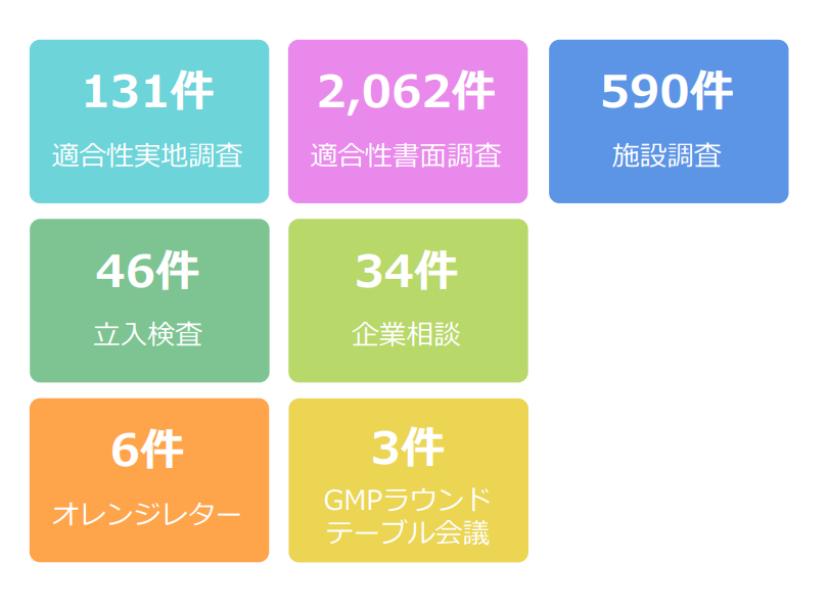
2024年,PMDA全年共实施了131件GMP/GCTP实地检查(“調査”),2,062件书面检查,以及590件设施检查和46件突击检查(“立入検査”)。此外,PMDA还处理了34件企业咨询,并发出了6封橙色警告信,组织了3次GMP圆桌会议,以促进行业内的交流和问题解决。
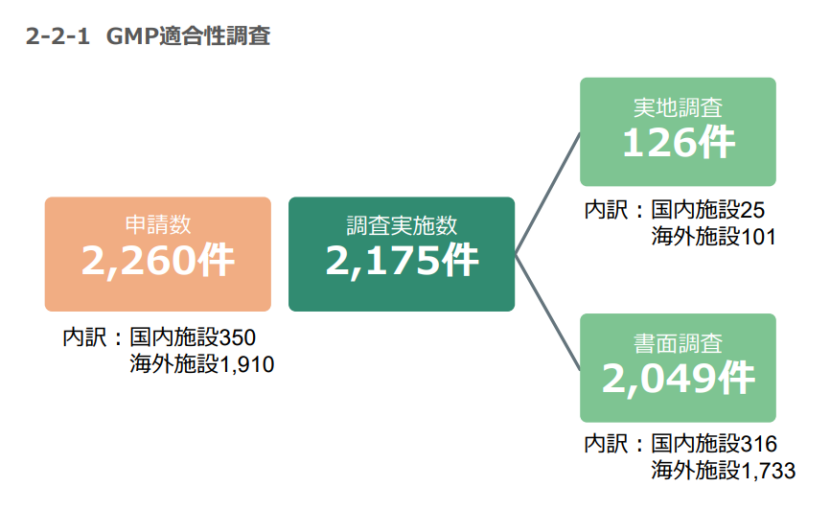
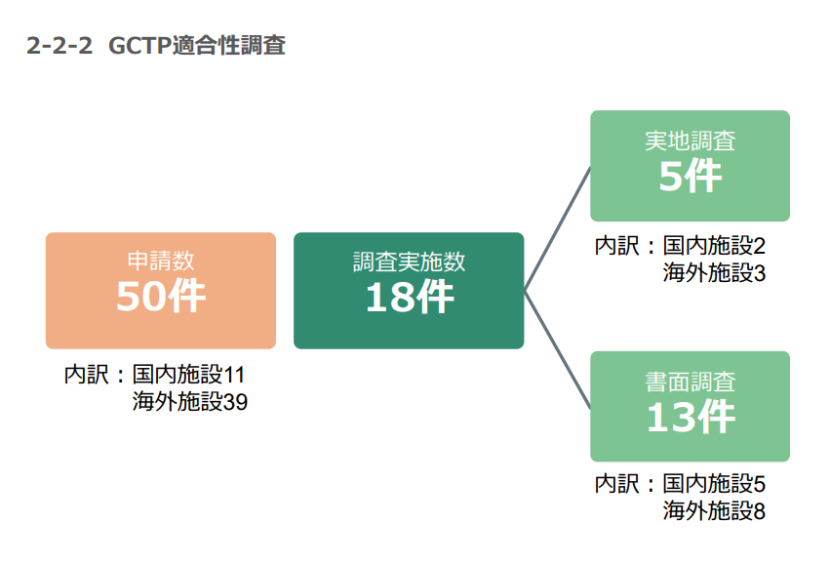
由上图可知,2024年,PMDA共收到2,260件需要GMP检查的申请,实际完成检查2,175件,大部分是通过书面完成的。在126件实地检查中,针对海外设施的检查远远超过国内设施。GCTP申请数量要少得多,且大多数申请并未实施检查。
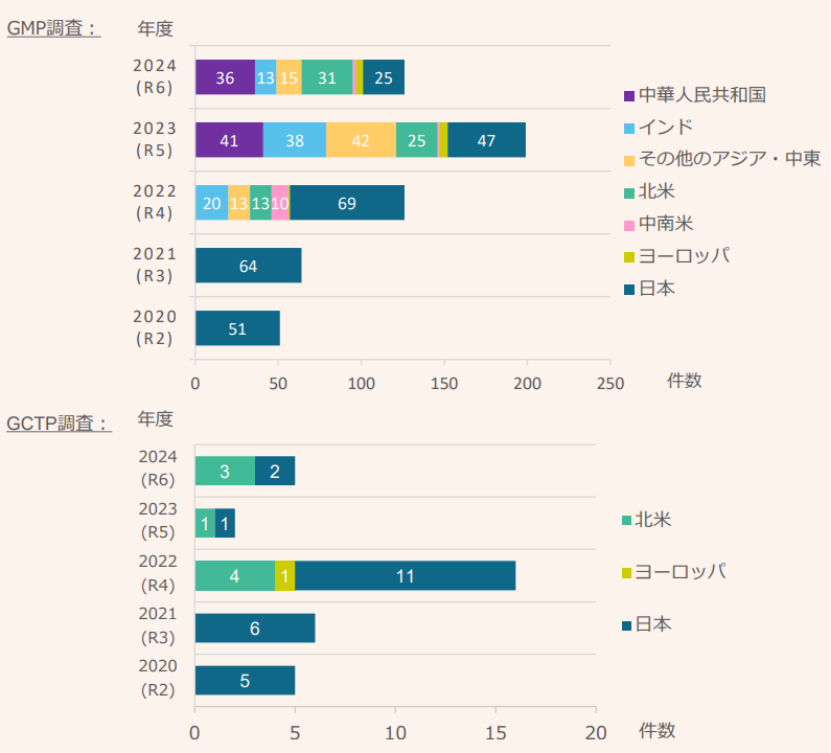
从检查的地域分布来看,2020年度和2021年度,由于新冠疫情导致各国实施了旅行限制,因此仅对日本国内场地进行了检查。2022年开始,海外场地检查开始恢复。2023年度重新开始对中国的实地检查,达到41件,与新冠疫情前持平。到了2024年,中国已成PMDA境外检查最多的目的地。另据报告,2024年度的实地检查数量与2023年度相比有所减少,但原因在于2023年补足了新冠疫情时未完成的工作,因此实际上2024年度的实地检查数量达到新高。
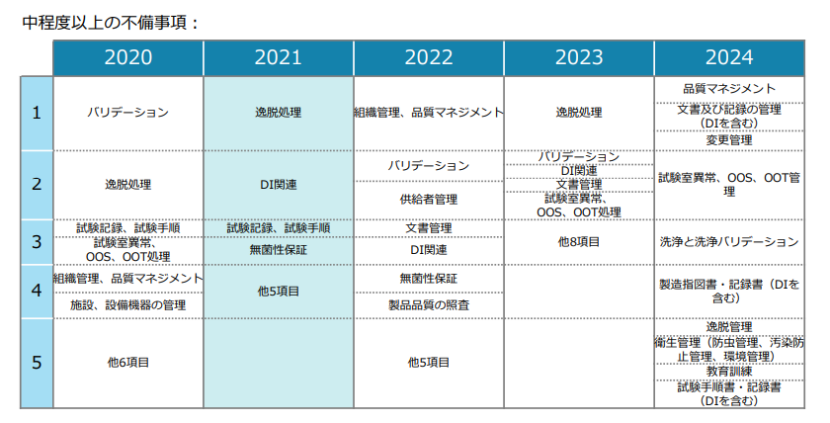
检查缺陷方面,中等程度以上的不合规项(包括中等和重大)在2024年有了比较明显的变化。主要集中在质量管理,文件及记录管理(包括数据可靠性DI),变更管理,实验室异常、OOS、OOT管理,清洁与清洁验证,生产指令与记录(包括DI),偏差管理,卫生管理(包括防虫管理、污染防止管理、环境管理),培训,以及检验规程与记录(包括DI)等方面。
突出缺陷在变更和偏差的沟通,但也有很基本的文件保存期限问题
PMDA专门分析了三类突出缺陷,集中在变更管理、偏差管理以及文件和记录管理方面。
变更管理方面,沟通问题严重。约半数的缺陷项涉及未明确规定与药品生产销售商(其职责类似于MAH)的沟通流程。部分企业仅评估变更对产品质量的影响,而未对变更对批准事项的影响进行评估,这可能导致变更未及时通知药品生产销售商。还有些企业错误地认为,只要变更不影响批准事项且无需进行药品监管事务手续,即使可能影响产品质量,也无需通知药品生产销售商。更有甚者,企业在与药品生产销售商沟通变更事项时,未规定记录沟通内容的流程。
在偏差管理方面也存在沟通问题。除了约一半的缺陷项涉及对个别偏差原因调查不足,与变更管理缺陷类似的是,约20%的缺陷项涉及未明确规定与药品生产销售商的沟通流程。部分企业仅评估偏差对产品质量的影响,而未对偏差对批准事项的影响进行评估。还有些企业将偏差是否通知药品生产销售商的判断权交给偏差管理负责人,但未明确规定具体的判断标准,导致重大偏差可能未被及时通知。
在文件和记录管理方面,最常见的缺陷项是文件和记录保存期限不足。根据日本GMP法规第20条规定,文件和记录的保存期限需遵循相关法规要求,例如生物制品和再生医疗产品等的文件和记录需在有效期基础上增加10年保存期限,但部分企业仅简单地将保存期限设定为10年。还有些企业错误地认为GMP法规规定的保存期限仅针对生产记录,而对其他需要保存的记录(如质量信息记录、设备清洁记录、原材料和产品出入库记录、试验设备使用日志等)保存期限较短。此外,有些企业在为日本市场批次和国外市场批次同时制备试验样品时,将制备记录归档到国外市场批次的记录中,导致实际保存期限未达到日本GMP法规要求的期限。
报告观察到,这些问题大多发生在海外生产企业,反映出海外企业对日本GMP理解不足。PMDA也在努力通过发布最新英文版GMP等方式,提高法规透明度。
识林-实木
识林®版权所有,未经许可不得转载
必读岗位: - QA:确保生产过程和质量控制符合省令要求。
- 生产:遵循省令指导,实施生产控制标准。
- 注册:了解省令对药品注册的影响,准备相应的注册文件。
适用范围:
本文适用于日本国内的药品和医药部外品,包括化学药、生物制品、疫苗和中药等。适用于所有在日本运营的药企,包括Biotech、大型药企、跨国药企、CRO和CDMO。 要点总结: - 生产控制标准: 明确了药品和医药部外品生产过程中必须遵守的质量控制标准。
- 质量控制要求: 强调了对药品和医药部外品的质量控制要求,包括原料、生产过程和最终产品的检验。
- 合规性审查: 规定了对生产控制和质量控制体系的合规性审查流程。
- 记录和报告: 要求企业保持完整的生产和质量控制记录,并在必要时向监管机构报告。
- 持续改进: 鼓励企业持续改进生产控制和质量控制流程,以提高药品安全性和有效性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |