首页
>
资讯
>
哪些更快“转正”?研究分析102项 FDA 肿瘤药加快批准
出自识林
哪些更快“转正”?研究分析102项 FDA 肿瘤药加快批准
2025-07-11
近日,一项发表于JAMA Network Open的研究《FDA 授予加快批准的抗癌药物获得完全批准或撤回所需时间的影响因素》 (Factors in Time to Full Approval or Withdrawal for Anticancer Medicines Granted Accelerated Approval by the FDA)剖析了FDA加快批准抗癌药物的全面批准或撤回时间的影响因素。
FDA的加快批准计划始于1992年,允许基于早期临床试验 中未验证 的替代指标(surrogate measures)来批准药物,前提是这些指标被认为可能合理预测临床获益。获得加快批准后,药企需进行确证性试验(confirmatory trials)以验证药物的实际临床获益。如果未能完成这些试验,药物可能会被撤回。
加快审批 的转正是否及时,如何确保确证性临床试验 有效开展,等等这些问题也将是我国监管和创新药企业接下来必须面对的重要课题。从FDA报告看 ,美国市场药企履行上市后承诺的情况颇为可观。NMPA则刚于7月7日再次征求《药品附条件批准上市申请审评审批工作程序(试行)》 意见,采取多种具体措施。
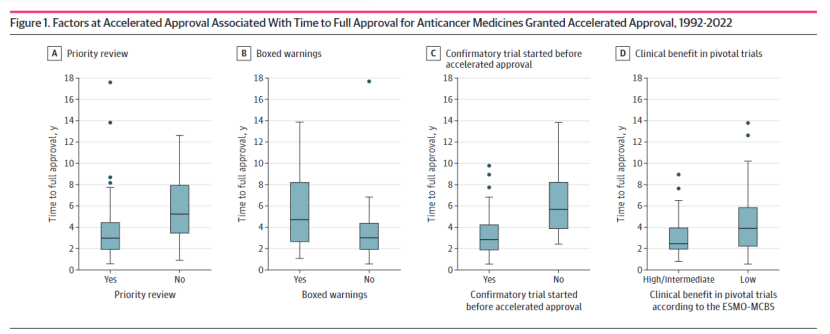
研究分析了1992年至2022年间获得FDA加快批准的102种抗癌药物适应症 ,这些药物在2024年8月31日之前已转换为全面批准。其中,81%的药物获得了优先审评 (priority review)资格,26%的药物在加快批准时带有黑框警告(boxed warnings)。结果显示,从加快批准到全面批准的中位时间为3.1年。
研究发现,加快批准时的几个关键因素与全面批准的时间显著相关。首先,获得优先审评资格的药物完成全面批准的时间显著短于未获得该资格的药物(中位时间分别为2.9年和5.1年,P=0.002)。其次,加快批准时已启动确证性试验的药物比未启动的药物更快完成全面批准(中位时间分别为2.78年和5.59年,P<0.001)。此外,加快批准时带有黑框警告的药物完成全面批准的时间更长(中位时间4.61年,不带黑框警告则为2.9年,P<0.001)。
在临床获益方面,欧洲医学肿瘤学会临床获益量表(ESMO-MCBS)被用于评估关键试验的临床获益。结果显示,加快批准时关键试验在ESMO-MCBS框架下显示中等或高获益的药物,完成全面批准的时间更短(中位时间2.37年,显示低获益则为3.81年,P=0.03)。确证性试验中显示总生存期 (overall survival)显著改善或生活质量(quality of life)显著提升的药物,其全面批准的时间也更短。
以上结论均符合监管和业界对于加快审评审批监管范式的良好预期。但研究也提出了几项政策建议,以优化FDA的加快批准路径。
例如,FDA应更加慎重考虑适用加快路径的药物。这些药物需具备有意义的活性水平、根据ESMO-MCBS定义的有意义的临床获益(在加快批准时至少具有中等获益),或是没有严重不良反应 的初始迹象。
此外,研究还敦促FDA利用其扩大的法定权力,严格执行现行法规指南,确保确证性试验在加快批准时已经开始或是完成招募,并每年公开报告确证性试验的状态。研究人员还给出建议,将支付方的报销与FDA确证性研究的截止日期挂钩,可能有助于确保确证性试验的及时完成。
识林-实木
识林® 版权所有,未经许可不得转载
适用岗位及工作建议:
RA(注册):必读。需熟悉修订后的医疗器械分类规则,以便在注册申报时准确分类医疗器械产品,确保合规性。 QA(质量管理):必读。应根据新规则更新质量管理体系,确保产品分类与监管要求一致。 R&D(研发):必读。在研发阶段即考虑医疗器械的分类,以指导产品设计和开发。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。