首页
>
资讯
>
对于细胞和基因治疗产品,与临床研究阶段相适应的GMP考量
出自识林
对于细胞和基因治疗产品,与临床研究阶段相适应的GMP考量
2022-07-01
细胞 和基因治疗 产品产业持续发展,从发现研究阶段逐步进入临床开发并过渡到中后期和及商业化生产阶段,产品的质量、合规以及适当的标准成为需要考虑的必要要素,到了是否准备好开始GMP 生产的决策点。
在着手考虑这些要素的组织或机构,包括成熟的大/中型生物药企业、初创公司、以及学术研究实验室等,在早期生产时经常问及这样的问题:“我们准备好开始GMP生产了吗?”,“1期产品生产适用的GMP是什么?”等。
Trevor Deeks在其《Phase Appropriate GMP for Biological Processes》书中给出了如下描述:“阶段相适应的GMP不仅仅是关于临床试验 中使用的药品的GMP期望,它还涉及在开发阶段实施适当的药品质量体系(ICH Q10 )以及该阶段所需的活动。还应考虑每个阶段的监管档案要求以及其与GMP、质量体系和文件记录的关系要求。”[1]
法规要求与相关指南
21 CFR Part 210.2(c) 规定“用于临床1期试验的研究用药品,如§312.21(a) 所述,需遵循21 U.S.C 351(a)(2)(B)的法规要求。该类药品的生产可豁免本章211部分的要求。”
但是,临床试验物料 受制于FD&C法案第501(a)(2)(b)节 的法规要求,其中规定,如果药品和器械不符合cGMP,则应视为掺杂 。FD&C法案501(a)(2)(b) [21 U.S.C. 351(a)(2)(B)]-掺杂药物和器械,“若其为药品,其生产、加工、包装 或贮存 所使用的方法、设施或控制不符合或没有根据cGMP 操作或管理,以确保此类药品符合本章关于安全性的要求,以及其声称或表明拥有的鉴别、规格、质量和纯度特征。
2008年发布的FDA行业指南:1期临床研究用药品cGMP ,清楚的介绍了相关法规及考虑背景,并指明了合适的实践方向,帮助企业建立适当的体系和程序,从而确保1期临床研究用药品适当的应用cGMP。
此外,WHO 和EU 均已发布相关指南,我国也在前不久发布《药品生产质量管理规范—临床试验用药品附录(试行)》 。
阶段相适应cGMP一般性考量
在项目进程中,用以支持1期研究产品的新建设施、改造设施或选择委托CDMO等决策,都可能因项目需要发生改变,比如需求调整和可用产能变化都可能影响这一决策。
有经验的CDMO通常会提供既定的质量体系以及就绪的标准。但申办方应积极参与,并负责确保已建立阶段相适应的控制水平,以及后续的开发。具有广泛经验的全新视角通常是进行“就绪能力的评估”的最快和最有效的方法,这对大多组织都有帮助。
在评估什么是阶段相适应的cGMP 时,申办方应当考虑1期产品生产用物料 的危害,产品剂型 相关背景,并记录这些危害和风险评估,以及风险缓解方法。规程的充分性和明确性,以及设备和环境的控制,是应用cGMP的基础。物料的输入和来源可能会在整个生命周期 中发生变化,应当适当受控,因为有些是关键的。同样,生成的数据的重要性也是至关重要的,生产和检验数据及记录应当符合数据可靠性 原则,需要进行充分的培训和认真的应用,加深对数据可靠性原则的理解。
在进行“就绪能力”评估时,应考虑包括但不限于:质量部门的设置及职责应明确并有文件记录;应由有经验和经过培训的人员根据现行规程开展活动;生产工艺及其规程等应经过验证;质量保证 要素,包括变更 、偏差 和CAPA 的管理、产品质量回顾 等。这些活动均应明确并用文件记录。
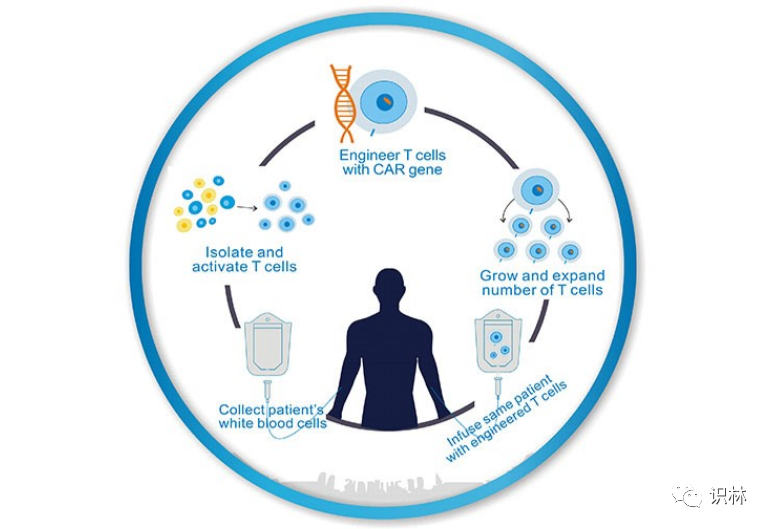
细胞和基因治疗等生物产品特殊方面考量
细胞库 对于基因治疗 和其它生物制品 的重要性值得特别考虑。用于生产质粒的病毒和细菌细胞库的制备应在GMP条件下进行,并且至少有一个主细胞库(MCB) ,其完全符合生产和外来因子的监管标准。可以在以后制备工作细胞库(WCB) 以确保生产的连续性,并且可以建立MCB和WCB的生长和生产率的可比性。
对于整个研发生命周期的进展,中间体和质粒的工艺应通过3期进行表征并得以验证。随着工艺能力的提高,质量标准 可能会随着时间的推移而调整,例如,在较早阶段,宿主细胞蛋白或宿主细胞DNA的含量应计划为2-5%,通过工艺改进或优化将其降低至更具代表性的1%的水平。
应提供用于确定安全性、鉴别、规格/效价、纯度和质量的合格分析方法 ,用于检验1期产品用物料 。必须对认证为无TSE/BSE的外来因子和原材料进行安全性检测。在第2/3期继续使用合格的分析放法,并且必须在3期工艺验证 之前完成分析方法的验证。宿主细胞蛋白或宿主细胞DNA残留的商业化试剂盒可在开发早期使用,但可能需要在生命周期后期和商业化时进行专门开发。分析仪器/设备应经过确认以开始GMP 检测。
设施和生产设备应经过规划确认,重点放在生产环境控制和关键设备上。设计确认很难在事后进行,因此,如果设备和设施计划在1期之后仍使用,最好在设计阶段完成确认;至少,设备和设施应针对预期用途进行调试、校准和维护。使用一次性设备和组件可以简化操作,但必须仔细管理采购和交付周期。灭菌工艺必须经过验证,并成功完成无菌确认,并进行操作员培训和确认。随着产品走向商业化,清洁规程和清洁记录必须到位,并进行清洁验证 。计算机系统必须符合其预期用途,并具备适当的数据可靠性 原则和控制措施。
参考资料
[1],Phase-Appropriate cGMP Considerations for Cell and Gene Therapy - Readiness to begin GMP manufacturing is a decision point that requires careful consideration. Keith Lamb, Executive Director, Lachman Consultants 05.02.22.
Deeks, T. Phase Appropriate GMP for Biological Processes, Chapter 2, "What is Phase Appropriate GMP ? –Regulatory Background and Current Expectations." ISBN: 978-1-942911-17-3, DHI Publishing, 2018.
识林-枍
识林® 版权所有,未经许可不得转载
适用岗位:
QA(质量保证):必读。负责确保所有制造过程符合CGMP要求,并监督质量控制功能。 研发(R&D):必读。需要理解CGMP在药物研发阶段的应用,特别是在I期临床试验中。 注册(Regulatory Affairs):必读。必须了解CGMP要求,以便在IND申请中正确提交CMC信息。 生产(Production):必读。负责实施CGMP规定的制造控制措施。 工作建议:
QA:确保所有I期研究用药品的生产活动遵循CGMP,并监督质量控制流程的有效性。 研发:在I期临床试验中应用CGMP,确保产品质量和安全性。 注册:在IND申请中包含符合CGMP要求的CMC信息,确保监管合规。 生产:根据CGMP要求制定和执行生产流程,确保I期研究用药品的生产质量。 适用范围:
文件要点:
CGMP合规性: 强调了I期研究用药品必须遵守CGMP规定,以确保药品的安全性、身份、强度、质量和纯度。人员资质: 规定了所有人员应具备执行其分配功能的教育培训和经验。质量控制功能: 明确了QC功能的角色和责任,包括材料审查、生产程序审批、批次放行或拒绝以及调查意外结果或错误。设施和设备控制: 强调了制造环境应有足够的工作区域和适当设备,以及适当的环境控制。特殊制造情况: 特别提到了多产品设施、生物和生物技术产品以及无菌产品/无菌加工产品的制造控制。以上仅为部分要点,请阅读原文,深入理解监管要求。
法规指南解读:ICH Q10 Pharmaceutical Quality System
适用岗位(必读):
QA:确保质量体系符合ICH Q10要求,监控质量体系的实施和持续改进。 注册:理解ICH Q10对药品注册的影响,确保注册文件与质量体系要求一致。 研发:在药品开发阶段应用ICH Q10原则,确保产品和流程的质量。 生产:根据ICH Q10要求,管理商业化生产过程中的质量控制和持续改进。 药物警戒:利用ICH Q10框架下的知识管理和质量风险管理,优化药物警戒活动。 工作建议:
QA应定期审查和更新质量手册,确保其反映ICH Q10的要求。 注册人员应确保所有注册文件和提交材料遵循ICH Q10的质量体系框架。 研发团队应将ICH Q10的原则整合到药品开发的每个阶段。 生产部门应依据ICH Q10建立和维护商业化生产的质量控制体系。 药物警戒部门应使用ICH Q10提供的工具进行风险评估和管理。 文件适用范围:
要点总结:
质量体系模型 :“ICH Q10提供了一个基于ISO质量概念的综合性药品质量体系模型,与ICH Q8和Q9相辅相成。”管理责任 :“高级管理层有最终责任确保有效的药品质量体系的建立,以实现质量目标。”持续改进 :“ICH Q10鼓励使用科学和基于风险的方法,在产品生命周期的每个阶段促进持续改进。”知识管理与质量风险管理 :“知识管理和质量风险管理是实施ICH Q10并成功实现其目标的推动因素。”监管方法 :“ICH Q10的实施效果通常可以在生产场所的监管检查中评估。”以上仅为部分要点,请阅读原文,深入理解监管要求。