首页
>
资讯
>
天时地利人和都不占,临门一脚失去加速批准资格
出自识林
2024-01-24
美国 FDA 于 2024 年 1 月 8 日发布了其 2023 年度 FDA 新药审批总结报告 ,其中的附录 B 列出了获批新药获得的认定及是否在首轮审评获批。从表格中能看出,绝大多数新药都有 1-5 项认定(首创、孤儿药 、快速通道、突破性治疗、优先审评 、加速批准 ),在 FDA 资源倾斜下,未能在首轮审评获批的品种更加引人注目。在逐个查看下列品种收到的完全回应函 (Complete response letter, CRL)时,发现 Elfabrio 先前不仅获得了表中列出的快速通道和优先审评认定,还获得过加速批准资格。那么最后的总结报告中为什么没标注呢?
Elfabrio (pegunigalsidase alfa (PRX102); BLA 761161)[1] 是由 Protalix 公司通过其专有的ProCellEx® 植物细胞蛋白表达系统生产的重组α-半乳糖苷酶-A稳定化蛋白,作为法布雷病(Fabry Disease, FD)的一种酶替代疗法 (enzyme replacement therapy, ERT)。
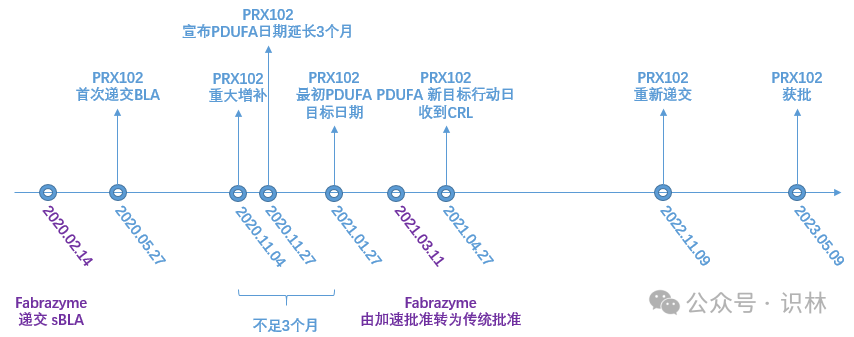
Chiesi 作为申请人 于 2020 年 5 月 27 首次递交 PRX102 的 BLA 申请,基于 PB-102-F01/02 (F01/02) 研究(单臂 、开放标签 ),主要终点是通过 BLISS 方法进行肾脏 Gb3 包涵体评分评估表明肾小管周围毛细血管 (PTC) 中肾脏 Gb3 包涵体较基线有大幅且具有统计显著性的降低(一种替代终点 ),寻求加速批准。彼时,FDA 只批准过两款法布雷病药物,2003 年获得加速批准的ERT疗法 Fabrazyme (agalsidase beta) 和 2018 年获得加速批准的 Galafold (一种蛋白质伴侣,FD 患者亚组)。
失去“天时”
在 PRX102 首轮审评过程中,同是 BLA 的 Fabrazyme (agalsidase beta) ERT 疗法,于 2021 年 3 月 11 日由加速批准转为传统批准 (2020 年 2 月 14 日递交 sBLA,PDUFA 目标日期是 2021 年 3 月 14 日)。这发生在 PRX102 BLA 行动日 2021 年 4 月 27 日之前,当考虑批准其它的治疗法布雷病的药品且想要通过加速批准途径时,就需要考虑 Fabrazyme 这种可用疗法了。因此,在没有足够的证据确定 PRX102 比现有治疗为患者提供了有意义的治疗益处的情况下,PRX102 不再符合加速审批的资格。
而查阅资料发现,在 2020 年 11 月 27 日,Protalix 和 Chiesi 称 FDA 将 PRX102 的 PDUFA 行动日期由 2021 年 1 月 27 日延长 3 个月至 2021 年 4 月 27 日。[2]
是什么原因导致了 PDUFA 目标日期的延长?如果 PRX102 的目标行动日期不延长,仍然是 2021 年 1 月 27 日,这时 Fabrazyme 还未转为“传统批准”,PRX102 能否顺利获得加速批准?
失去“人和”
通过查阅 FDA 公布的 Elfabrio 审评卷宗,发现可能是导致 PDUFA 目标日期延长的原因:提交的数据与原始文件不一致。
FDA 科学调查办公室(OSI)在对 F01/02 研究的有效性数据对照原始文件副本进行交叉检查时,发现原始文件与提交的 Gb3 终点数据之间存在一些差异,另外,OSI 审评员指出 2 个受试者的读数分数间存在很大差异。OSI 审评员建议申办者确认所有场地、所有受试者的 BLISS 分数之后提交修订后的数据集。
2020 年 11 月 4 日,申请人提交了修订后的有效性数据,而这引发了重大增补 。根据 21 CFR 314.60(b) ,在至首次审评周期末不足 3 个月递交重大增补资料,审评周期将延长不超过 3 个月。
失去“地利”
如果申办者在数据递交前能够做到更严谨的处理数据集,是否能赶在目标日期前获批?
答案也是否定的。
Chiesi 在 2021 年 4 月 27 日收到完全回应函 ,通读审评卷宗中披露的完全回应函,其指出两项缺陷:①在生产设施 进行的记录审查中发现问题,②因可用疗法而造成的加速批准 的可用性问题(见前文描述)。
此外还因旅行限制无法对位于以色列的生产场地进行批准前检查 ,而这是批准前必须解决的事项且期限是未知和不可控的。
“意外”并不意外
在 2022 年 11 月 9 日重新提交时,申请人递交了 PB-102-F20 (F20) 研究,寻求 PRX102 的传统批准。F20 是一项随机 、双盲 、Fabrazyme 阳性对照研究,在 77 名成年 FD 受试者 中按 2:1 随机分配至 PRX102 组或继续使用 Fabrazyme,主要疗效终点是 2 年的年化肾小球滤过率估算值 (Estimated Glomerular Filtration Rate, eGFR) 的变化(斜率)。
而 F20 这项 Ⅲ 期研究早在 2015 年 11 月 3 日 Ⅱ 期研究结束会议上作为主题被提出,研究开始时间为 2016 年 6 月[3] ,在 2019 年 2 月 27 日 Type C 会议上拟议确证性临床试验 为正在进行的 F20 研究。
但是 FDA 对于 F20 的结果,接受得也没有那么顺利。
在重新递交前,2021 年 4 月,申办方根据 12 个月的数据对 F20 试验进行了揭盲中期分析,中期分析是在 protocol 中写明的以支持在 EMA 的递交。2021 年 9 月 9 日,在审评结束会议上讨论针对 CRL 问题的回复,申请人提出将 F20 的主要评估治疗 24 个月后 PRX-102 相对于 Fabrazyme 的优效性 改为非劣效性 (NI)2 /年,与基于 12 个月数据的中期分析所用的值相同。FDA 认为,计划评估 2 年时间的 PRX102 相比于 Fabrazyme 在 eGFR 斜率上的非劣效性可能是一种合理的方法,前提是有足够的理由和证据支持这种统计方法,但未能接受申请人提出的裕度值。
最终,审评小组确定 F20 研究不能支持非劣效性主张,因为缺乏数据支持 Fabrazyme 的非劣效性裕度。给力的是,FDA 为了帮助解释 F20 研究中两个治疗组之间 eGFR 斜率结果,并提供有关 Fabrazyme 在研究人群中的预期效果的信息,审评组考虑了其它外部数据评估检测敏感性问题。虽然这样的评估存在明显的局限性,但是这些信息有助于将 F20 试验结果结合起来并得出结论,PRX-102 和 Fabrazyme 之间的 eGFR 终点的可比结果提供了支持性的证据。F01/02 中评估的肾脏 Gb3 终点不是临床终点 ,因为它不能直接测量患者在日常生活中的感觉、功能方面的影响、或者患者的生存时间。但是基于发表的文献可证明药物对该终点的影响具有临床相关性。虽然两项临床研究 均存在局限性和不确定性,当综合考虑并结合其它学科信息时统计团队认为 PRX-102 的有效性是有实质性证据的。
确立PRX102 对 Fabry 患者有效的实质性证据包括:
F20 研究结果表明,经过两年的研究产品暴露后,接受过 ERT 治疗的患者随机接受 PRX102 或 Fabrazyme(一种已批准的具有相同作用机制的 ERT)患者之间的年化 eGFR 斜率相当。
PRX102 对疾病特异性生物标志物 的药理作用(未接受过 ERT 治疗的患者血浆 Lyso-Gb3 水平降低)。
o 该疾病的公认病因是由于单一酶缺乏导致的鞘糖脂代谢的单基因先天性错误。
o PRX102 作为内源酶缺陷/缺失的外源酶替代物的靶向作用机制。
Elfabrio 最终于 2023 年 5 月 9 日获得传统批准。
法布雷病 & Protalix
法布雷病是一种 X 染色体连锁遗传病,基因缺陷致使溶酶体 α-半乳糖苷酶活性缺陷,导致一种称为三酰基神经酰胺 (Gb3) 的脂肪物质在人体溶酶体中逐渐异常沉积。Gb3 的异常储存随着时间的推移而增加,因此,Gb3 主要在血管和组织中积聚。Gb3 沉积的最终后果包括疼痛发作和外周感觉受损到终末器官衰竭。每 40,000 至 60,000 人中就有 1 人患有法布雷病。
Protalix 是第一家获得FDA批准的通过基于植物细胞的悬浮表达系统生产的蛋白质的公司。Protalix 独特的表达系统代表了一种以工业规模开发重组蛋白的新方法。Protalix 与 Chiesi 合作在美国境内外开发和商业化 pegunigalsidase alfa。[2]
参考资料
[1] Elfabrio (pegunigalsidase alfa (PRX102); BLA 761161) 审评卷宗
[2] 2.0 2.1 Protalix BioTherapeutics and Chiesi Global Rare Diseases Announce U.S. Food and Drug Administration Acceptance of a Resubmitted Biologics License Application for Pegunigalsidase Alfa for the Proposed Treatment of Fabry Disease
[3] 【NCT02795676】Study of the Safety and Efficacy of PRX-102 Compared to Agalsidase Beta on Renal Function (BALANCE)
作者:识林-樟
识林® 版权所有,未经许可不得转载