首页
>
资讯
>
国会鼓吹降价对创新影响不大?美国版“医保国谈”的坎坷之路
出自识林
国会鼓吹降价对创新影响不大?美国版“医保国谈”的坎坷之路
2022-07-21
上个月,2022年度国内医保谈判大幕拉开,又将迎来新一轮“灵魂砍价”。大洋对岸的美国,药价也早已成为舆论焦点 ,困难重重。正巧近日美国Medicare医保谈判也有新动态,借此机会来梳理一下此事的前因后果。
Medicare是美国的联邦健康保险计划,面向65岁及以上人群,残疾者,终末期肾病患者以及渐冻症患者。Part A住院保险覆盖医院、专业护理机构等住院期间用药;Part B医疗保险通常覆盖患者无法自行给药的药品,比如静脉输注的肿瘤药物;Part D处方药物福利计划,覆盖从零售药店或邮购药房获取的药品;Part C优势计划有多种多样的计划可选,在覆盖Part A和Part B的同时提供其他福利,大多数也包含处方药福利。
根据现行法律,美国政府无法直接与制药公司谈判干涉Medicare的药价。民主党迄今为止已有3次尝试意图颠覆这个规则,然而一直以来十分坎坷,屡屡碰壁。最早可追溯到the Elijah E. Cummings Lower Drug Costs Now法案(简称H.R.3),于2019年在众议院通过,但是后续参议院并未对其进行表决,在2021年重新引入众议院,此次却止步于能源和商业委员会。2021年众议院通过了The Build Back Better法案(简称BBBA) ,包括气候变化,家庭救助和药价管控等条款,在参议院有机会走预算和解程序以简单多数票(51票)通过,即在50名共和党参议员全部反对的情况下,需要获得所有民主党参议员外加副总统的支持。然而民主党参议员曼钦因担忧通货膨胀和国债公开表示不会支持该法案,掐灭了该法案通过的希望。就在上周,一份意在重振BBBA的新草案公开,聚焦在药价和医保,获得了所有50名民主党参议员的支持,有望绿灯通过。
虽一再缩水,但政府医保谈判一旦落地,具有里程碑意义
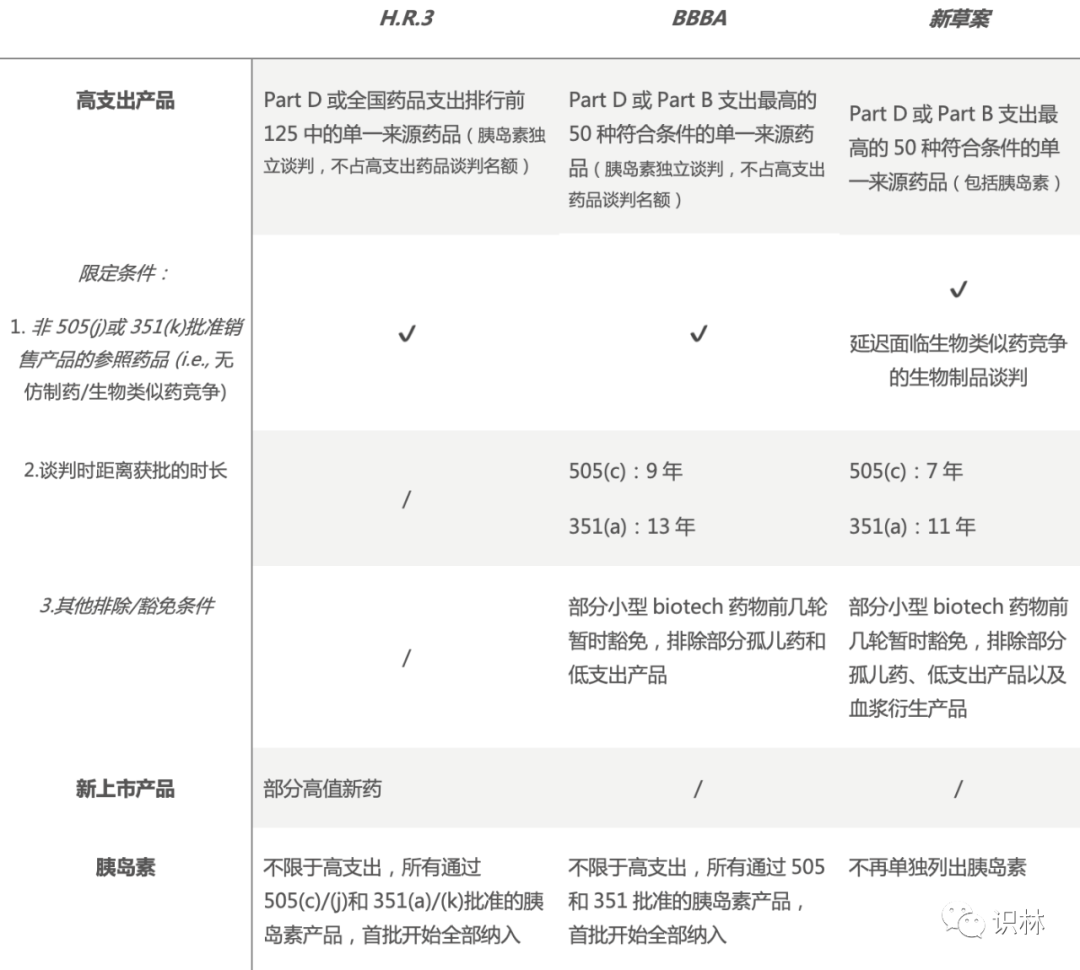
立法过程向来艰难,法案往往历经数次修改,BBBA最初版的药价谈判相关条款跟H.R.3基本一致,而后一再缩水,下文中主要对各法案的最新版本进行比较。各法案针对药价部分都包括了Medicare药价谈判,涨价过快超通胀率的药品需要支付回扣等条款。药价谈判流程相似,正常是在谈判价格实行前2年开启谈判,新草案中提出首批谈判需再提早一年,谈判设立最高公平价格(Maximum Fair Price, MFP),定价的规则有所不同,不同意谈判的制造商会被征收高昂的特种商品消费税(Excise tax)。相较于H.R.3的谈判价格适用对象覆盖范围广,还包括了商保,如今BBBA和新草案仅适用于Medicare。
符合谈判资格的药品条件方面,共同点是针对的药品来源单一,高支出产品均需尚未面临仿制药 或生物类似药 的竞争。H.R.3纳入的药品范围最广,可以覆盖部分高值的新上市产品,而BBBA和新草案针对的是成熟产品,并针对药品距离获批不同的时长设定了不同的MFP天花板。值得注意的是,新草案针对部分很可能在谈判价生效日前获批并销售的生物类似药的生物制品 施以延迟谈判。具体内容对比可见下表:
注:505指《联邦食品、药品和化妆品法》第505节 ;351指《公共卫生服务法》第351节
根据新草案,2026和2027年第一批10种和第二批15种Part D药品的谈判价格正式生效,2026年起(即第三批)开始Part B药品的谈判。除非因为新增适应症 等原因获得重新谈判的资格,协议一直持续到药品不再符合谈判要求,比如仿制药 上市,首次谈判后每年的MFP在前一年MFP的基础上按城市居民消费价格指数的年度百分比增幅增加,更多细节请参阅原文件:
url ;
2021.09版本:url
政府谈判到底是否损害药品创新?企业总是挂在嘴边,国会真给算了笔账
到底应不应该通过Medicare药价谈判和其他直接的药价管控措施,支持反对两方的争论焦点在于这些政策导致的收入减少是否会显著降低制药公司的研发投入,进而使得未来“无药可用”。
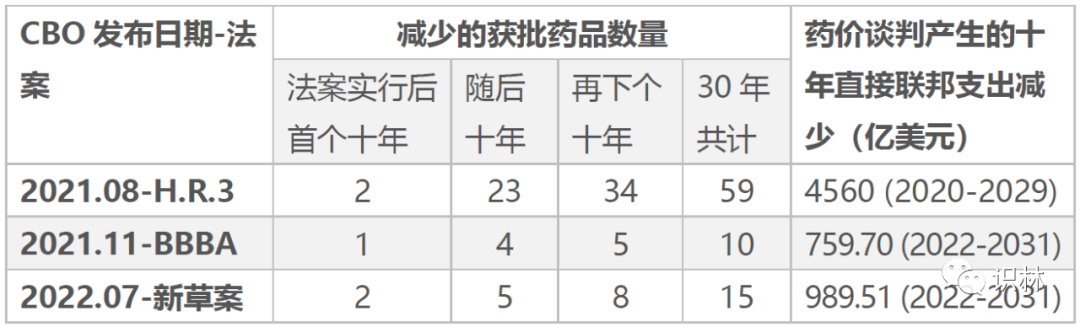
国会预算办公室(CBO)相继对3项法案进行成本估算以及新药获批影响估计,如下表所示。
细究创新层面,大趋势是,因为药品开发耗时很长,短期影响较小。相较于H.R.3,由于谈判药物的选择更加受限,且定价方式不同,BBBA和新草案的药价管控政策对创新的影响更加有限。“国谈”之后10年内,不过是少批2个创新药?听着还真是令人放心呢。
CBO的估算方法发生过几轮迭代和修改,作为基线的年获批药品数量从原本的30提升到44,反映2015-2019年间平均每年获批药品的数量。从基于文献估计创新弹性(elasticity of innovation)转向为制药公司的投资决策过程建模,模型最开始仅覆盖临床1期到3期几个关键节点,在每个节点上根据开发成功的可能性,预期的收益是否超过开发成本决定是否继续推进,后又新增政策对临床前开发决策和加速批准 产生的影响,以及是否会驱动小型公司更高的资本成本。
虽经过数次改进,但是估算所基于的研究不确定性仍然很大,也不乏有对其过分简单化决策过程,数据选择和假设不合理的批判,比如假设公司在各节点决定是否继续推进只考虑了单个产品,而忽视了投资决策往往取决于公司的整个管线,又比如将作为基线的年获批药物数量设定为恒定值忽视了技术发展可能带来的增速,无法反映现实,因此有声音提醒这个模型单用于学术交流是很有意思,但是要用于支撑政策则需谨慎。
药价管控是否会阻碍创新,是否会为科学进程踩下刹车,其影响大小众说纷纭。现有的证据不够有说服力,研究的假设各有千秋,难有完美的模型考虑到方方面面。这些“损失”的药对公众健康会造成多大影响也难以定夺,要么,假设它们都是潜在的救命药,不要侥幸,“一个都不能少”;要么,考虑不同药物临床价值各有不同,很多低价值药物可能掺杂其中,而这个风险经过权衡后是可以承担的?至于对策,药价管控导致的R&D支出减少能否通过加大科研资金的投入弥补 ,又要多少才能填上窟窿?除了新药,药价谈判是否会导致仿制药 和生物类似药 竞争弱化也有待研究。
现在的可负担和未来的有药可用只能二选一吗?何以兼得是个问题。
参考资料
1. url
2. url
3. url
4. url
5. url
识林-梣
识林® 版权所有,未经许可不得转载
适用岗位: 必读岗位:注册专员(负责药品注册文件的准备与提交)、研发人员(涉及新药开发与研究)、临床研究协调员(负责临床试验的监管与协调)、QA(确保药品生产过程符合法规要求)、市场准入专员(涉及药品市场策略与准入)。 工作建议: 注册专员:确保所有新药申请文件符合Sec. 355的要求,特别注意专利信息的提交与更新。 研发人员:在新药开发过程中,及时与注册部门沟通,确保研究数据支持药品安全性与有效性。 临床研究协调员:监督临床试验过程,确保数据的完整性和合规性,及时报告关键信息。 QA:在药品生产和质量控制过程中,遵循Sec. 355的规定,确保产品质量。 市场准入专员:了解新药的市场准入要求,包括专利挑战和市场独占期等。 适用范围: 本文适用于在美国进行注册的化学药品和生物制品,包括创新药和仿制药。发布机构为美国食品药品监督管理局(FDA),企业类别包括Biotech、大型药企和跨国药企。
要点总结: 新药申请批准的必要性 :强调了新药上市前必须获得FDA的有效批准。申请文件内容要求 :明确了新药申请需包含的详细内容,如安全性、有效性研究报告,成分列表,制造过程描述等。专利信息与挑战 :规定了新药申请中专利信息的提交要求,以及对专利有效性或侵权的认证和通知程序。批准流程与时间限制 :详述了FDA审批新药申请的流程,包括审批时间限制和听证机会。药品安全性与有效性的持续监管 :强调了即使药品批准后,若发现新的安全性问题或缺乏实质性证据,FDA仍有权撤销批准。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读建议:
生物制品研发人员(R&D) :需关注生物制品的定义、开发标准和许可要求。质量保证专员(QA) :应确保生物制品的生产、加工、包装和储存符合安全、纯净和有效的标准。注册专员(Regulatory Affairs) :负责提交生物制品许可申请,了解审批、中止和撤销生物制品许可的要求。市场准入专员(Market Access) :需了解有关生物制品市场独占期的规定,以便制定市场策略。临床研究协调员(Clinical Research Coordinator) :负责确保儿科研究的合规性,并在适当时进行儿科研究。文件适用范围:
要点总结:
生物制品许可要求 :明确了生物制品进入州际商业必须持有生物制品许可,并满足特定标记和标签要求。设施检查与产品召回 :授权官员在合理时间内进入和检查任何生物制品的生产设施,并在产品对公共健康构成紧急危害时,规定了产品召回的程序和相关处罚。生物制品定义与许可 :对生物制品进行了定义,并规定了生物制品作为生物类似药或可互换产品的许可标准。专利和市场独占期 :详述了与生物制品相关的专利问题处理流程,以及生物制品的市场独占期规定,包括儿科研究对市场独占期的影响。法律效力与其他法律的关系 :阐明了本章与《联邦食品、药品和化妆品法》等其他法律的关系,并规定了生物制品许可的法律效力。以上仅为部分要点,请阅读原文,深入理解监管要求。