首页
>
资讯
>
PHSS 发布污染控制策略(CCS)指南文件集,详解如何准备和实施 CCS
出自识林
PHSS 发布污染控制策略(CCS)指南文件集,详解如何准备和实施 CCS
2023-08-02
药物与医疗保健科学协会(PHSS)近期发布了一份 146 页长的污染控制策略指南文件集 ,汇集了开发和实施污染控制策略 所涉及的各个方面,包括准备 CCS 的指导和模版,以及相关风险分析:影响与关键性分析(FMECA)、故障模式影响以及污染控制的关键性分析。
自欧盟 GMP 附录 1 修订以来,污染控制策略(CCS)的要求显然是强制性的,并且不太可能改变,因此,PHSS CCS 聚焦小组工作开始考虑附录 1 对 CCS 的要求、范围和内容以及在文件监管框架中的定位。
GMP 和无菌药品附录 本身已经涵盖了无菌药品生产中污染控制策略的很多内容,ICH Q9 质量风险管理 和 ICH Q10 质量体系 也在企业有了较广泛的应用,所以污染控制策略并非是一个全新的要求,其“新”之处只在于要求企业专门针对污染控制,整合已有的控制措施,建立一个全面的整体方案,以确定和连接整个生产体系中的关键控制点 (Critical Control Point,CCP),并评估与污染 风险相关的所有控制措施(设计、流程、技术和人员)和监测措施的有效性。更多有关污染控制策略的知识可点击进入“污染控制策略 ”主题词阅览。
CCS 的“连接”还旨在提供所有支持现场文件的连接,这些文件分别设计要素(设施 /工艺/程序)、确认、控制、监控和相关工艺风险管理。工艺风险管理包括一系列风险评估,涵盖与预防、污染检测和应对管理相关的过程。
PHSS 通过各种培训和会议平台讨论制定 CCS 的目的、内容和结果。PHSS 将这些讨论和会议结果生成了 CCS 文件集,其中包括准备 CCS 的指南和模版。
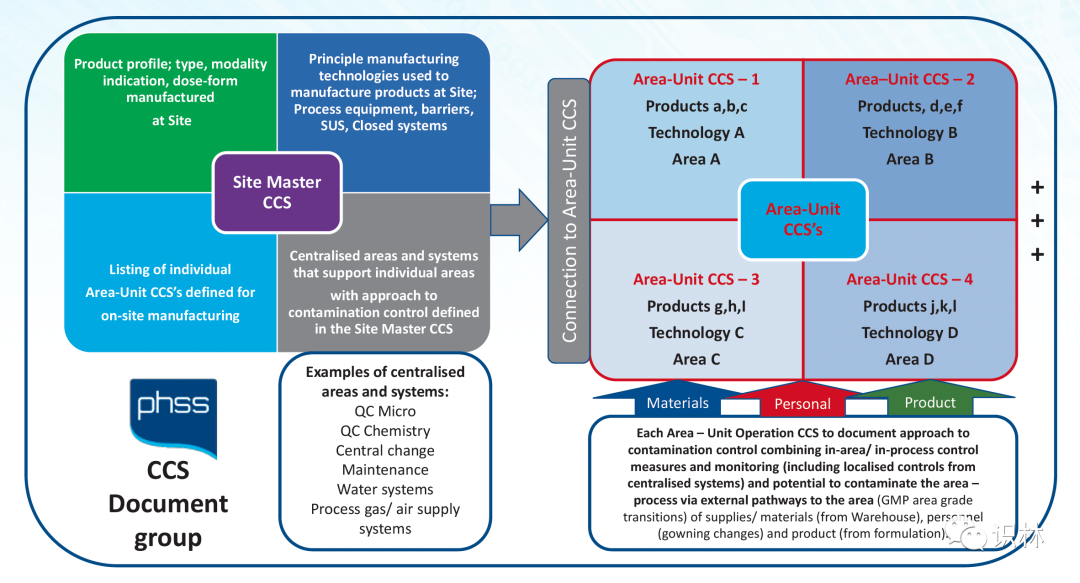
PHSS 表示,从一开始就没有把 CCS 视为一个单一的文件,而是一系列相互关联的 CCS 文件的集合;包括单一的场地主 CCS 和涵盖不同的产品组/生产技术的多个区域(单元操作 )CCS,每个单独的 CCS 在产品生产中都有独特的污染风险概况。场地主 CCS 被认为与场地主文件 交叉引用,从而连接到文件的监管框架中。
可以考虑将场地主 CCS 和区域 CCS 的元素结合起来的单一 CCS 用于单一场地生产产品,这类产品的生产技术具有明确的污染风险概况和相关的应用控制和监测措施以减轻风险。
PHSS CCS 指南文件集提供了 CCS 准备的起点,包括:
准备 CCS 的 SOP ,考虑在多个场地或单一场地采用统一方法。
为场地主 CCS 中的每个内容标题准备 CCS 时要考虑的要点。
准备 CCS 区域中每个内容标题时需要考虑的要点。
用于执行污染控制 FMECA 风险分析以支持 CCS 准备的 SOP。
Excel FMECA 工具,根据附录 1 和修订版 ICHQ9(R1) 的要求完成 FMECA 风险分析,考虑产品生产中污染的关键性和严重性。该 FMECA 的特点是限制主观性,是 ICHQ9(R1)的关键关注点和要点。
文件集给出了 PHSS CCS 文件结构和链接图示:
识林-椒
识林® 版权所有,未经许可不得转载
法规指南解读
适用岗位 :
工作建议 :
QA:确保所有生产活动符合GMP要求,监督无菌药品生产流程。 生产:按照GMP要求执行无菌生产操作,确保产品质量。 研发:在药品开发阶段考虑GMP合规性,设计符合要求的生产流程。 临床:确保临床试验用药的无菌性和质量符合GMP标准。 注册:在药品注册过程中提供符合GMP要求的生产和质量控制信息。 适用范围 :
要点总结 :
无菌药品生产环境 :强调了对无菌药品生产环境的严格控制,包括洁净室的分类和设计,以及对生产环境的持续监测。质量风险管理(QRM) :在整个文件中,QRM是确保无菌药品生产质量的核心原则,要求企业在设计和控制生产设施、设备、系统和程序时应用。关键控制点 :提出了无菌药品生产过程中的关键控制点,包括设施设计、设备操作、过程验证、环境监测和人员培训等。污染控制策略(CCS) :强调了CCS在无菌药品生产中的重要性,要求企业实施全面的CCS以确保产品质量和安全。无菌工艺验证 :要求对无菌工艺进行验证,包括无菌过程模拟(APS)和其他相关测试,以确保生产过程能够持续产生无菌产品。以上仅为部分要点,请阅读原文,深入理解监管要求。
法规指南解读:ICH Q10 Pharmaceutical Quality System
适用岗位(必读):
QA:确保质量体系符合ICH Q10要求,监控质量体系的实施和持续改进。 注册:理解ICH Q10对药品注册的影响,确保注册文件与质量体系要求一致。 研发:在药品开发阶段应用ICH Q10原则,确保产品和流程的质量。 生产:根据ICH Q10要求,管理商业化生产过程中的质量控制和持续改进。 药物警戒:利用ICH Q10框架下的知识管理和质量风险管理,优化药物警戒活动。 工作建议:
QA应定期审查和更新质量手册,确保其反映ICH Q10的要求。 注册人员应确保所有注册文件和提交材料遵循ICH Q10的质量体系框架。 研发团队应将ICH Q10的原则整合到药品开发的每个阶段。 生产部门应依据ICH Q10建立和维护商业化生产的质量控制体系。 药物警戒部门应使用ICH Q10提供的工具进行风险评估和管理。 文件适用范围:
要点总结:
质量体系模型 :“ICH Q10提供了一个基于ISO质量概念的综合性药品质量体系模型,与ICH Q8和Q9相辅相成。”管理责任 :“高级管理层有最终责任确保有效的药品质量体系的建立,以实现质量目标。”持续改进 :“ICH Q10鼓励使用科学和基于风险的方法,在产品生命周期的每个阶段促进持续改进。”知识管理与质量风险管理 :“知识管理和质量风险管理是实施ICH Q10并成功实现其目标的推动因素。”监管方法 :“ICH Q10的实施效果通常可以在生产场所的监管检查中评估。”以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位及工作建议 QA(质量保证) :应深入理解质量风险管理的原则和工具,确保公司的质量管理体系符合ICH Q9(R1)的要求。研发 :在药物开发阶段应用质量风险管理,以建立对风险场景的知识和理解,确保商业制造阶段的适当风险控制。生产 :在生产过程中实施质量风险管理,以识别和控制潜在的质量风险,确保产品质量和供应的连续性。注册 :在药品注册过程中,利用质量风险管理数据和分析来支持注册申请,展示产品的风险控制措施。文件适用范围 本文适用于化学药、生物制品和生物技术产品的质量风险管理,包括原料药、创新药、仿制药和生物类似药。适用于全球范围内的Biotech、大型药企、跨国药企、CRO和CDMO等不同企业类别,由ICH发布。
文件要点总结 质量风险管理定义 :“质量风险管理是一个系统的过程,用于评估、控制、沟通和回顾药品质量的风险。”风险管理方法 :介绍了多种风险管理工具,如FMEA、FMECA、FTA、HACCP等,以及它们在药品质量风险管理中的应用。风险管理过程 :强调了风险评估、风险控制、风险沟通和风险回顾的重要性,并提供了相应的步骤和方法。风险管理的整合 :讨论了如何将质量风险管理整合到行业和监管操作中,以及如何通过培训和教育提高行业和监管人员对质量风险管理的理解和信心。产品可用性风险 :特别强调了质量/生产问题对产品供应的影响,以及如何通过质量风险管理来预防和减轻由此产生的风险。以上仅为部分要点,请阅读原文,深入理解监管要求。
法规指南解读:欧盟GMP
适用岗位(必读):
QA:确保质量体系符合GMP要求,监控文件管理及生产过程。 生产:遵循GMP规定,保证生产过程的合规性。 研发:在药品开发阶段考虑GMP要求,确保可生产性。 注册:了解GMP对药品注册的影响,准备相关文件。 药物警戒:监控药品安全性,确保符合GMP对药品监控的要求。 适用范围:
要点总结:
制药质量体系(Chapter 1): 强调建立和维护全面的制药质量体系,确保产品质量和合规性。人员(Chapter 2): 规定了人员资质、培训和卫生要求,以保证生产过程的人员符合性。厂房设施与设备(Chapter 3): 明确了生产环境和设备的GMP要求,以防止交叉污染和保证生产条件。文件管理(Chapter 4): 规定了文件的编写、审核、批准和存档要求,确保生产和质量控制的可追溯性。无菌药品生产(Annex 1): 更新了无菌药品生产的GMP要求,包括对特定条款的实施期限的调整。以上仅为部分要点,请阅读原文,深入理解监管要求。