首页
>
资讯
>
FDA拟全面更新医用气体认证、GMP、和上市后等生命周期监管法规体系
出自识林
FDA拟全面更新医用气体认证、GMP、和上市后等生命周期监管法规体系
2022-06-07
5月23日,美国FDA发布一项拟议法规(Proposed Regulation),将对若干医用气体 (Medical Gas)的制定认证法规,修订对cGMP 、上市后安全报告及标签 的要求,现向业界开放90天的意见征询,8月22日截止评论。(查看原文内容详情 )
这项拟议法规将为医用气体的药品制造法规新增加两个部分,21 CFR Part 213和230(目录请详见下文);相应的,对关联的法规进行修订更新,如21 CFR Part 201 、210 和211 等。如果最终确定,该拟议规定将澄清制造、加工、包装、标签或分销医用气体的实体的监管法规义务。
拟议法规的主要内容举例
标签的要求。澄清成分和含量 声明,修订若干认定医用气体的警告声明,并为散装或运输容器建立更加限制性的标签要求。
cGMP 的要求。建立专门针对医用气体的cGMP法规,21 CFR Part 213。该法规包括与Part 211 诸多相同类别的条款,但认识到并反映医用气体 在制造、标记和发运方面的重要差异,包括容器和标签 的重复使用,定稿后,将代表医用气体的cGMP最低要求;
■不同的清洁要求,因为气体通常在密闭/封闭的加压系统中制造,而且在不适当的时间清洁可能会引入污染物;
■不同的时限要求,不规定生产中的时间限制,因为医用气体通常不会过期或降解;
■容器和密封系统新增要求,除与Part 211.94 中相同的一般要求外,新提出供个体患者使用的便携式低温医用气体容器和小型低温气体容器应具有工作仪表,用以指示容器内是否具有足够的气体以供继续使用。
■回收重新利用建议。与常规药物cGMP法规回收要求不同,贮存不当的医用气体可以回收,除非不利条件对产品的鉴别、规格、质量或纯度,或产品容器密封完整性 产生不利影响。
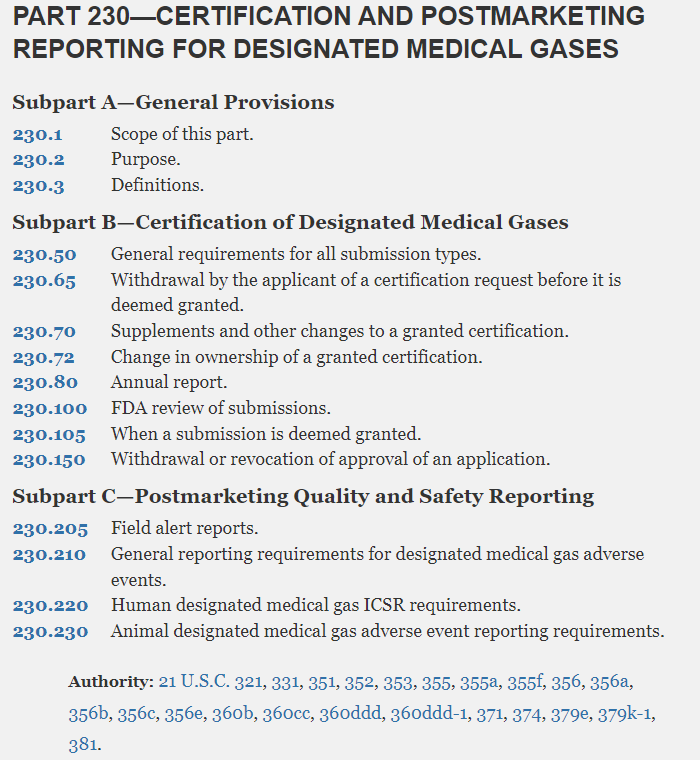
拟议将认定医用气体认证流程编入法规,包括关于补充申请 、年报 以及撤回或撤销批准申请的规定。
上市后安全报告的要求的例外情况。对于使用氧气时的患者或者动物死亡相关事件无需向FDA报告,除非申请人或非申请人确有证据表明氧气与死亡之间存在因果关系。解释是向处于危急状态的人或动物提供氧气时,死亡是预期内的结局,除非有明确证据,FDA认为此类报告不太可能反映潜在的安全信号或不会对氧气安全性带来新的认识。
Part 210和211中拟定的修订和更新
在Part 210.1 和Part 210.2 中增加对Part 213的引用,以便Part 210 中的使用条款反映新增的医用气体cGMP法规。
在Part 211 中范围中增加Part 213的引用,并删除正文部分中有关医用气体 要求的描述。Part 211.1(a) 中增加“medical gases as defined in § 213.3(b)(12)”,删除Part 211.94(e) ,删除Part 211.125(c) 的最后一句医用气体描述,删除Part 211.132(c)(1) 中的参考“containers of compressed medical oxygen”,删除Part 211.170(b) 中描述“Reserve samples of compressed medical gases need not be retained”,删除Part 211.196 中描述“For compressed medical gas products, distribution records are not required to contain lot or control numbers”。
拟议增加的Part 213和230章节目录
21 CFR Part 213 - Current Good Manufacturing Practice for Medical Gases
医用气体现行生产质量管理规范(拟新增)
21 CFR Part 230 – Certification and Postmarketing Reporting for Designated Medical Gases
认定医用气体的认证和上市后报告(拟新增)
美国其他医用气体法律法规指南
FD&C Act Part G - Medical Gases (Sections 360ddd - 360ddd-2)
FDA 医用气体cGMP指南 (2017.06)
FDA 2016医用气体容器-密封件规定 问答 (2017.01)
FDA 认定医用气体认证流程 (2015.11)
FDA 压缩医用气体指南 (1989.02)
识林用户可登录,参考主题词【医用气体(Medical /Medicinal Gas)】
参考资料
1. Current Good Manufacturing Practice, Certification, Postmarketing Safety Reporting, and Labeling Requirements for Certain Medical Gases
2. Current Good Manufacturing Practice, Certification, Postmarketing Safety Reporting, and Labeling Requirements for Certain Medical Gases (Proposed Rule) Preliminary Regulatory Impact Analysis
作者:识林-枍
识林® 版权所有,未经许可不得转载。
适用岗位:
必读岗位:QA(质量保证)、注册(Regulatory Affairs) QA工作建议:确保公司生产的医用气体容器和标签符合FDA的最新CGMP规定,特别是关于容器颜色和标签的具体要求。 注册工作建议:监控法规变化,更新注册文件,确保公司遵守新的标签和容器规定。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
解读法规指南
适用岗位:
QA(质量保证部门) 注册部门 市场准入部门 研发部门 临床研究部门 工作建议:
QA:确保所有产品标签符合21 CFR 201的规定,监控标签的变更和审核流程。 注册部门:在提交药品注册资料时,包括标签设计,确保其符合FDA的标签要求。 市场准入部门:在市场推广材料中使用标签信息时,确保信息的准确性和合规性。 研发部门:在药品开发阶段考虑标签要求,以确保最终产品的标签合规。 临床研究部门:确保临床试验中的药品标签正确反映了试验要求。 适用范围:
要点总结:
标签信息要求: 强调了药品标签上必须包含的信息,如活性成分、使用说明、警告、剂量形式和强度等。净含量声明: 规定了药品标签上净含量的表达方式,包括重量、度量或计数,并要求准确反映包装内药品或设备的数量。警告和注意事项: 要求药品标签上必须有清晰的警告和使用上的注意事项,以确保患者安全使用。特殊人群用药: 对孕妇、哺乳期妇女、儿科和老年用药进行了特别规定,要求标签上提供相应的用药信息和警告。药品相互作用: 要求标签上必须包含可能的药品相互作用信息,以帮助医疗专业人员和患者了解潜在的风险。以上仅为部分要点,请阅读原文,深入理解监管要求。
岗位必读指南:
QA(质量保证):确保生产过程符合cGMP规定,保障药品质量与安全。 生产:遵循cGMP标准进行药品生产、加工、包装或储存。 QC(质量控制):进行药品质量检测,确保符合规定标准。 适用范围:
要点总结:
cGMP规定的地位 :强调了cGMP规定是确保药品符合安全、身份、强度、质量和纯度要求的最低标准。合规性与监管行动 :不遵守cGMP规定的药品将被视为掺假,相关责任人将面临监管行动。HCT/Ps的额外要求 :对于HCT/Ps,除了cGMP规定外,还需遵守特定的捐赠者资格和适用的当前良好组织实践程序。适用性与冲突解决 :cGMP规定与其他相关法规相互补充,如有冲突,特定适用于药品的法规优先。临床研究药品的豁免与合规 :I期临床研究药品生产可豁免部分cGMP规定,但进入II期或III期临床研究或合法上市后,必须符合cGMP。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位必读指南:
QA:负责确保所有操作符合cGMP要求,包括生产、质量控制、设备维护等。 生产:必须遵守书面程序,确保产品质量。 质量控制(QC):负责样品的测试和批准或拒绝,以及稳定性测试。 设备维护:确保设备清洁、维护和校准符合规定。 仓储与分销:遵守药品存储和分发的书面程序。 文件适用范围:
文件要点总结:
质量控制单元的责任: 必须有一个质量控制单元,负责批准或拒绝所有组件、药品容器、包装材料、标签和药品,并审查生产记录以确保没有错误发生或错误已得到全面调查。
人员资质与责任: 参与药品生产、加工、包装或储存的人员必须具备相应的教育、培训和经验,并遵守良好的卫生习惯。
设备设计、清洁与维护: 设备应适当设计,便于操作、清洁和维护,并按规定进行定期清洁和维护。
组件和药品容器的控制: 必须有书面程序详细描述组件、药品容器和闭合件的接收、识别、存储、取样、测试和批准或拒绝。
生产和过程控制: 必须有书面程序确保药品具有其声称或代表的身份、强度、质量和纯度,包括偏差处理和产量计算。
包装与标签控制: 必须有书面程序确保正确的标签和包装材料用于药品,包括防篡改包装要求。
仓储与分销程序: 必须有书面程序描述药品的存储和分发,确保药品质量。
实验室控制: 必须建立科学合理的规格、标准、抽样计划和测试程序,以确保药品及其组件符合适当的身份、强度、质量和纯度标准。
记录与报告: 所有与生产、控制或分发相关的记录必须保存至少一年,或在特定情况下保存更长时间,并随时可供授权检查。
退回和报废药品的处理: 退回的药品必须被识别并保留,除非证明其符合适当的安全、身份、强度、质量和纯度标准,否则应销毁。
以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位及工作建议:
QA(质量保证):必读。确保生产过程符合cGMP要求,审核生产记录。 注册:必读。熟悉标签和市场后安全报告要求,准备相关文件。 生产:必读。遵循cGMP生产医用气体,确保产品质量。 药物警戒:必读。负责收集和评估上市后安全数据,及时报告。 适用范围:
文件要点总结:
cGMP合规性: 明确要求医用气体生产商遵守现行生产质量管理规范(cGMP),确保产品质量和安全。认证要求: 规定了医用气体生产商的认证流程,强调了对生产设施和过程的审核。上市后安全报告: 强调了生产商对上市后安全数据的收集、评估和报告责任,要求及时向FDA报告严重不良事件。标签要求: 详细规定了医用气体产品的标签内容,包括适应症、剂量、警告等信息,以确保患者和医护人员了解产品特性。质量控制: 新增了对医用气体质量控制的要求,包括原料检验、生产过程监控和成品放行标准。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
工作建议:
QA:审查和更新质量管理体系,确保符合新的CGMP要求,特别是针对医用气体的变更。 生产:调整生产流程,以适应标签重用、路边填充书面许可的灵活性等新规定。 法规事务:深入理解新的法规要求,为公司提供合规指导,特别是在标签要求和市场后安全报告方面。 适用范围:
要点总结:
CGMP成本节约:提出了针对医用气体的CGMP特定要求,预期通过简化检查流程和减少不必要的设施要求来节省成本。 标签要求变更:要求在氧气最终使用容器上增加“禁止吸烟”和“禁止电子烟”的警告,以减少火灾风险。 认证过程明确:明确了市场指定医用气体(DMGs)的认证请求流程,提高流程透明度。 市场后安全报告:对DMGs的市场后安全报告要求进行了修改,明确了必须报告的不良事件。 潜在成本节约:通过移除或放宽不适用于医用气体的CGMP要求,可能带来额外的成本节约。 以上仅为部分要点,请阅读原文,深入理解监管要求。
【文件概要】
【适用范围】
【影响评估】
【实施建议】
必读岗位: 生产/工程 :需确保设备设计符合气体专用性要求(如专用歧管),并建立容器检查(如死环测试、气味检测)和真空处理程序。 QC/QA :制定组件和成品气体测试计划,验证供应商分析报告可靠性,完善实验室记录以证明非药典方法的等效性。 注册 :确认企业及产品已完成FDA注册与列名,更新操作程序以纳入指南允许的替代性合规措施。 HRC运营 :明确转移操作中的标签责任,建立车辆储罐气体测试记录系统。 以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
QA(质量保证部门):负责确保所有操作符合CGMP要求,监控质量体系的实施。 生产(Production):负责按照CGMP要求进行药品生产,确保生产过程的合规性。 实验室控制(Laboratory Controls):负责药品的取样和测试,确保药品符合质量标准。 工作建议:
QA:定期审查和更新质量协议,确保与供应商的沟通机制有效,以及所有CGMP相关的操作都有书面程序和记录。 生产:在生产过程中,严格执行设备校准、清洁和维护程序,确保所有操作符合CGMP要求。 实验室控制:确保所有测试方法经过验证,并且测试结果准确记录,以支持药品的质量保证。 适用范围:
文件要点总结:
以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位:
QA:确保公司生产和储存的医疗气体符合官方药典标准。 注册:关注法规变化,及时更新注册文件和策略。 研发:在开发新医疗气体时,考虑官方药典标准和新药申请的相关规定。 适用范围:
文件要点总结:
医疗气体定义: 明确了医疗气体包括氧气、氮气、一氧化氮、二氧化碳、氦气、一氧化碳和医用空气等,必须符合官方药典标准。指定医疗气体: 新增了由卫生部部长根据新药或新兽药申请情况确定的其他医疗气体。法规修订要求: 规定了卫生部部长需在一定时间内评估并报告是否需要对联邦药品法规进行修订。最终法规发布: 如果确定需要修订,卫生部部长需在规定时间内发布最终法规。规则解释: 明确了本法规不适用于2012年5月1日前批准的药品,以及某些特定情况下的气体和组合。以上仅为部分要点,请阅读原文,深入理解监管要求。
适用岗位:
RA(注册) :必读。需了解医疗气体的认证流程、标签要求及处方要求,以便在注册过程中符合FDA规定。QA(质量管理) :必读。需掌握医疗气体的认证标准和标签要求,确保产品质量合规。研发 :必读。在开发医疗气体产品时,需遵循FDA的认证要求和使用指示。适用范围:
文件要点总结:
医疗气体认证流程 :明确了医疗气体认证的申请流程和所需信息,包括气体描述、赞助商及生产设施信息等。认证后的监管 :获得认证的医疗气体被视为满足某些FDA要求,但仍需遵守所有适用的批准后要求。标签要求 :指定医疗气体的标签需包含特定信息,如警告声明和存储处理指南。处方要求 :一般情况下,指定医疗气体需遵守处方药要求,但FDA有权豁免。特定用途的氧气可免处方使用。认证撤销 :若发现认证申请中存在重大遗漏或虚假信息,FDA可撤销认证。以上仅为部分要点,请阅读原文,深入理解监管要求。
必读岗位建议:
QA(质量保证):应关注医疗气体的合规性,确保不收取不适用的费用。 注册(Regulatory Affairs):需了解医疗气体的注册分类和费用豁免规定,以便在注册过程中正确应用。 适用范围:
文件要点总结:
医疗气体费用豁免: 明确指出,根据21 U.S.C. 360ddd-1条款被视为具有批准申请的指定医疗气体,无论是单独还是与其他指定气体组合使用,均不得根据21 U.S.C. 379h(a)或379j-12(a)条款收取费用。法律修订记录: 2016年的修订增加了对379j-12(a)条款的引用,扩大了豁免范围。以上仅为部分要点,请阅读原文,深入理解监管要求。