|
首页
>
资讯
>
FDA希望标准化PQ-CMC数据和术语
出自识林
2017-07-11
美国 FDA 于 7 月 10 日表示正在起草和征询关于使用标准化药品质量/化学制造和控制(PQ/CMC)数据元素用于电子提交的意见。
FDA 正在考虑作为 HL7(Health Level 7)结构化产品标签(SPL)文件实施 PQ/CMC 要求,表示,标准化 PQ/CMC 数据元素和术语的举措将提高药品审评过程的效率和质量。这一转变是根据《2012 食品药品管理局安全与创新法案》(FDASIA)完成的工作的一部分。
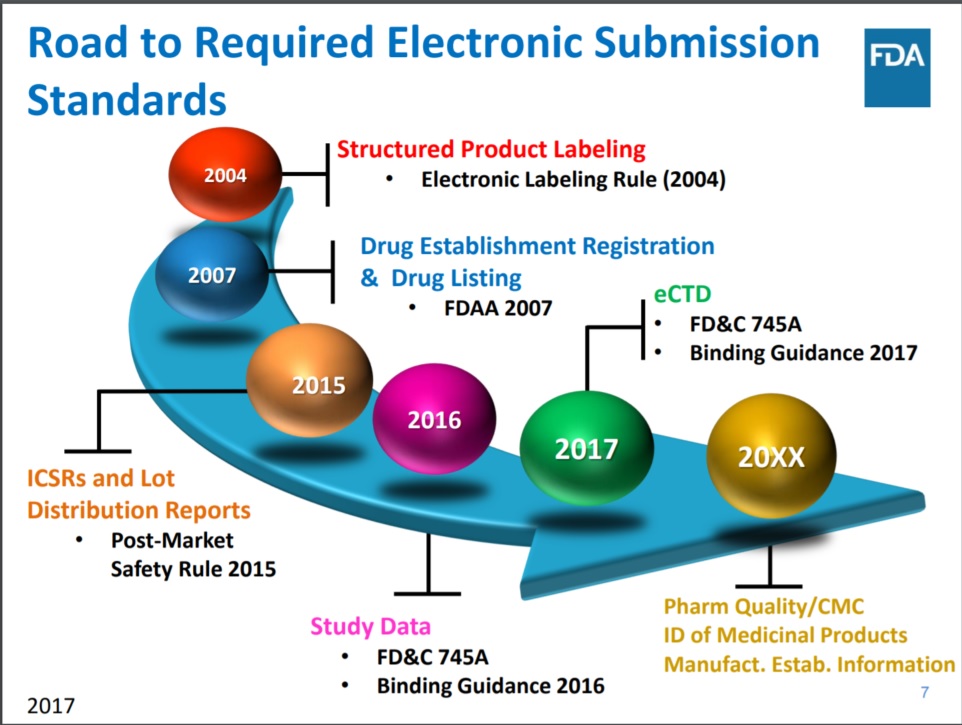
在评议方面,FDA 希望听取业界对 PQ/CMC 提交的数据元素和术语的准确性、适应性和适当性的意见。根据 FDA 战略计划办公室高级顾问 Ron Fitzmartin 近期在 2017 PharmaSUG 会议上演讲幻灯片 的更新,FDA 要求电子提交标准的路径如下: 的更新,FDA 要求电子提交标准的路径如下:
- 2004 年 结构化产品标签(2004 电子标签规定)
- 2007 年 药品设施登记和药品入录(2007 FDAA)
- 2015 年 个体案例安全性报告(ICSRs)和批分销报告(2015 上市后安全规定)
- 2016 年 研究数据(FD&C 745A;2016 年系列指南)
- 2017 年 eCTD(FD&C 745A;2017 年系列指南)
- 20XX 年 药品质量/CMC;医药产品识别信息;制造设施信息。
来源:Ron Fitzmartin 在 2017 PharmaSUG 会议的演讲幻灯片
FDA 表示,“PQ/CMC 数据结构化格式的开发将使 PQ/CMC 数据提交的内容和格式保持一致,从而为提交内容提供一种协调统一的语言,允许审评人员查询数据,并且有助于通过创建一个标准化的数据字典建立更高效和有效的监管决策过程。”
拟定的 PQ/CMC 数据元素和术语可在 www.regulations.gov 上获取(卷宗号 FDA- 2017-N-2166)或点击下载 pdf。
整理:识林-椒
识林®www.shilinx.com版权所有,未经许可不得转载。如需使用请联系admin@shilinx.com
参考资料
岗位必读建议: - 注册:了解FDASIA对药物和医疗设备注册审批流程的影响。
- 研发:关注创新药和仿制药的用户费用要求,以及对儿科药物开发的支持。
- QA:确保产品质量和安全性符合FDASIA规定的标准。
文件适用范围:
本文适用于美国境内的创新药、医疗设备、仿制药和生物类似药的注册分类,由美国食品药品监督管理局(FDA)发布,适用于Biotech、大型药企、跨国药企等各类企业。 文件要点总结: - 用户费用授权:FDASIA授权FDA从行业收取用户费用,以资助创新药、医疗器械、仿制药和生物类似药的审查工作。
- 儿科药物开发鼓励:该法案重新授权两个鼓励儿科药物开发的项目。
- PDUFA和MDUFA的第五次和第三次授权:这是处方药用户费用法案(PDUFA)的第五次授权和医疗器械用户费用法案(MDUFA)的第三次授权。
- 审查流程的稳定性和可靠性:通过这些用户费用计划,确保了审查人员队伍的稳定和审查流程的可靠性。
- 仿制药和生物类似药的用户费用计划:新计划建立在PDUFA和MDUFA成功的基础上,为仿制药和生物类似药的审查提供资金。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |