|
首页
>
资讯
>
APIC 发布原料药亚硝胺杂质风险管理指南
出自识林
APIC 发布原料药亚硝胺杂质风险管理指南
2025-06-02
5 月21日,原料药委员会(APIC)正式发布《亚硝胺风险管理指南:原料药生产商指南》第二版。该指南在 2020 年初版(Additional guidance on the assessment on the risk assessment for presence of N-nitrosamines in APIs)  基础上,结合最新监管要求(主要是EMA)与行业实践,系统更新了亚硝胺风险评估、检测与控制的全流程标准,为全球原料药(API)企业应对亚硝胺风险管理提供实操性框架。 基础上,结合最新监管要求(主要是EMA)与行业实践,系统更新了亚硝胺风险评估、检测与控制的全流程标准,为全球原料药(API)企业应对亚硝胺风险管理提供实操性框架。
修订指南增加了大量内容,全面讨论了亚硝胺的形成机制,将研究范围从单一的亚硝胺扩展到亚硝胺原料药相关杂质(NDSRIs);探讨了通过清除因子(Purge Factor,PF)计算进行理论风险评估的方法;提供了关于API测试(包括方法论和可接受限度计算)的风险详细评估指南;此外还讨论了如何将亚硝胺风险管理整合到质量管理体系中。
近两个月来,FDA、EMA、ICH、WHO多个机构陆续发布亚硝胺杂质可接受摄入限度、风险评估及预防控制的指南或更新,令药企目不暇接,丝毫不能松懈。
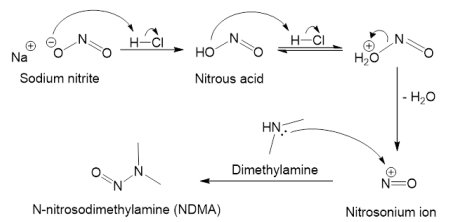
亚硝胺杂质的形成机制与风险来源
目前确定的亚硝胺杂质来源的风险因素复杂多样,包括但不限于:
- 亚硝化反应,当二级或三级胺和亚硝化剂在有利条件下反应时,可形成亚硝胺。比如作为亚硝胺前体的原料、中间体或API在酸性或高温等工艺条件下亚硝化;API被亚硝胺剂污染;使用受污染的回收物料(如溶剂、催化剂等);多用途设备的交叉污染;水中的亚硝酸盐与胺反应;离子交换树脂中的胺类渗出;空气中的氮氧化物(NOx)与二级胺反应等;
- 降解,由固有反应性(硝基烷基、肟或其它功能基团)或外源性亚硝化剂降解API产生;
- 包装,易产生亚硝胺杂质的包装主要与成品制剂相关,可认为该风险因素与API本身无关,可忽略不计。
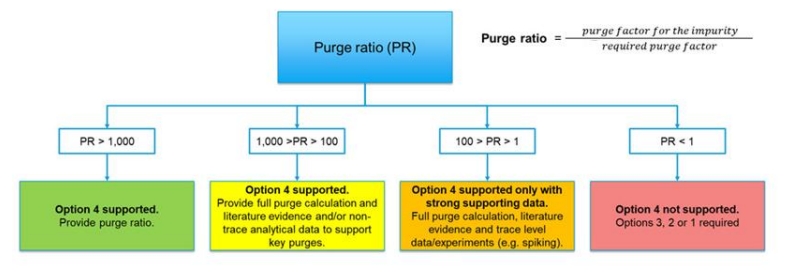
清除评估模型
在亚硝胺风险初步评估中,可采用基于ICH M7(R2)Option 4的清除评估,通过清除因子(PF)计算模型论证风险等级。该模型通过量化亚硝胺的关键物化性质(水溶性、挥发性、离子化特性等)与工艺步骤(如水洗、蒸馏)的清除能力,预测杂质去除效率。若计算所得总PF值超过所需清除因子(理论最大初始浓度/AI限值)1000倍以上,可判定风险可忽略,适用Option 4;若总PF值低于1-100倍,则表明存在潜在风险,需通过分析测试进一步验证(如图1)。该模型不仅优化了资源分配,还为企业工艺改进提供了科学依据,但需注意不同地区监管机构接受度差异。
API测试方法与限度设定规范
当风险评估提示API可能有亚硝胺风险时,要进行确认性测试或理论预测,参考EMA的MAH/申请人问答:关于人用药中的亚硝胺杂质(条款5(3)转介)的 CHMP 意见,指南详细介绍了API确认性测试的限度设定计算方法和试验方法。确认性测试需对一定数量的批次进行检测,根据结果采取不同控制策略,确保亚硝胺水平远低于可接受摄入限度。
质量管理体系的整合与优化
指南最后增设 "亚硝胺风险管理融入 QMS" 章节,其关键在于组织需提升员工风险认知,开展跨部门培训,并整合监管情报,安排团队收集解读法规;加强供应商管理,预审供应商并定期审计;实施全生命周期管理,把风险管控贯穿产品从研发到生产的全流程,根据变更调整策略。
识林-筱筱
识林®版权所有,未经许可不得转载
必读岗位及工作建议- MAH/申请人:负责确保药品中亚硝胺含量控制在规定限度内,设计或调整生产工艺以预防亚硝胺形成,并在必要时提交上市许可变更。
- QA:监督药品生产过程,确保符合GMP规范,审核亚硝胺风险评估和控制策略。
- 研发:在药品开发阶段考虑亚硝胺风险,参与制定和优化生产工艺。
- 注册:了解法规要求,准备和更新药品注册文件,包括亚硝胺风险评估和控制措施。
文件适用范围本文适用于所有人用药(化学药和生物制品),包括创新药、仿制药、生物类似药和原料药。适用于在欧盟上市的药品,由EMA发布,适用于Biotech、大型药企、跨国药企、CRO和CDMO等企业。 文件要点总结- 亚硝胺风险评估:所有人用药必须进行亚硝胺风险评估,无论市场状态或产品类型。
- 审查呼吁:针对化学合成API和生物API的药品,要求MAH进行风险评估、确认性测试和风险缓解措施。
- 风险缓解措施:MAH需设计生产工艺和控制措施,以预防或减少API和成品药中N-亚硝胺的存在。
- 限度设定:根据ICH M7(R2)原则,为亚硝胺设定特定的可接受摄入量(AI)限度。
- 监管要求:欧盟监管机构与国际伙伴合作,协调监管要求,确保药品质量和安全。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA(质量保证):负责确保原料药生产全过程符合质量管理规范,监控质量体系运行。
- QC(质量控制):负责原料药的质量检测,确保产品质量符合标准。
- 生产:负责按照GMP要求进行原料药的生产操作,确保生产过程合规。
- 工程:负责厂房设施和设备的维护保养,确保生产环境和设备符合要求。
适用范围:
本文适用于化学药领域的原料药生产,包括创新药和仿制药,适用于大型药企、跨国药企以及CRO和CDMO等企业类别,发布机构为国际通用标准。 文件要点总结:
原料药的生产质量管理规范强调了从质量管理到生产控制的全过程管理。首先,文件明确了质量管理的原则和机构职责,特别强调了质量保证和质量控制的重要性,并规定了自检、产品质量回顾以及质量风险管理的具体要求。在人员方面,规定了资质、培训和卫生要求,确保员工符合岗位需求。厂房与设施章节详细规定了设计建造、公用设施和特殊隔离要求,以保证生产环境的适宜性。设备章节则涉及设计建造、维护保养、校准和计算机化系统的要求,确保设备运行的可靠性。文件还特别提到了无菌原料药的生产特点,包括生产工艺、厂房设施设备设计、生产过程管理以及环境控制等,这些都是确保原料药质量的关键环节。 以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。
- 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。
- 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。
适用范围:
本文适用于涉及化学药、生物制品、疫苗和中药等药品类型的企业,包括创新药、仿制药、生物类似药和原料药等注册分类。适用于不同规模的企业,如Biotech、大型药企、跨国药企、CRO和CDMO等,由相关药品监管机构发布。 文件要点总结: - 质量管理体系概述:明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。
- 产品质量实现要素:涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。
- 质量保证要素:包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。
- 质量风险管理:介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。
- 质量管理系统文件:规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |