|
首页
>
资讯
>
2014年CDER新药审评概况
出自识林
2014-12-16 识林
作者:识林-Kapok
2014年CDER新分子实体计划运行平稳,突破性治疗药品成为亮点。新药申请提交数保持平稳,但35个新药获批,使得2014年成为FDA新药审批的大年。新药首轮获批通过率保持高位(74%)。罕见病用药获批15个,创下《罕见病用药法案》于1983年开始实施以来的新纪录。FDA批准的新分子实体药品继续领跑全球。3个新分子实体抗菌药品获批,为近年来所罕见。首个生物类似物药品正在审评之中。尽管成绩斐然,但面临的挑战依旧,这些挑战主要体现在新计划的实施和预期带来工作量增加,专才招聘和吸引、留住专才,以及在旅行和薪酬方案上受限。
在完成处方药生产商付费法案(PDUFA)规定的目标方面,FDA完成和超过该法案规定的几乎所有申请审评目标。全面实施PDUFA和《FDA安全与创新法案》(FDASIA)也为FDA提供了新的资源和竞争优势。FDA新药办公室在编人员数维持小幅增长,2013财年为916位全职雇员,计划2015年增加至975位,但这一数字仍然低于相关法案规定的1058位全职雇员封顶数。但离满足各项计划实施对人员配备的需求,存在一定距离。这些计划包括突破性治疗药品认定、生物类似物、以患者为关注点的药品研发(PFDD)、抗生素研发激励法案(The Generating Antibiotics Incentives Now (GAIN) Act)、仿制药生产商付费法案(GDUFA)、优先审评凭单等。联邦政府招聘体系、薪酬冻结、薪酬封顶和过时的薪酬体系,不利于招聘和吸引保留必需的专才。
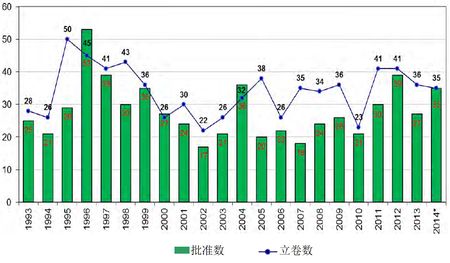
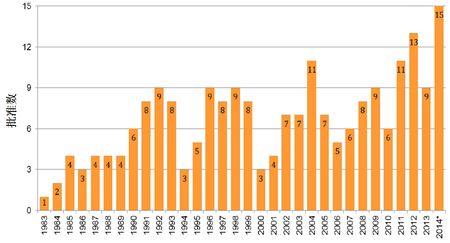
新药审批方面,处于研发中的新药IND管道形势喜人,增长动力主要来源于生物制品。截止2014年12月3日为止,2014财年CDER受理35项新分子实体药品申请(最近10年的平均受理数为34项)。截止2014年12月11日止,2014财年CDER批准35个新分子实体药品(最近10年的平均数为26个),其中突破性治疗药品7个(2013财年为3个),罕见病用药15个;罕见病用药获批数创下1983年《罕见病用药法案》实施以来的新高。首轮获批率保持高水平,但由于各项计划的执行,中位批准时间有所上升。
2013-2014财年CDER药品审评概览
|
| 2013财年
| 2014财年
|
| 申请类型
| 立卷数
| 绩效
| 立卷数
| 绩效
|
| 优先审评新分子实体NDA/原创BLA
| 17
| 100%
| 25
| 100%
|
| 标准审评新分子实体NDA/原创BLA
| 29
| 93%
| 15
| 100%
|
| 优先审评非新分子实体NDA
| 8
| 88%
| 9
| 89%
|
| 标准审评非新分子实体NDA
| 76
| 97%
| 71
| 100%
|
| 第I类NDA/BLA再提交
| 9
| 100%
| 7
| 100%
|
| 第II类NDA/BLA再提交
| 36
| 97%
| 31
| 100%
|
| 优先审评有效性补充申请
| 28
| 96%
| 37
| 100%
|
| 标准审评有效性补充申请
| 108
| 92%
| 131
| 99%
|
| 第I类有效性补充申请
| 1
| 100%
| 7
| 100%
|
| 第II类有效性补充申请
| 7
| 86%
| 8
| 88%
|
| 之前批准申请的生产补充申请
| 637
| 90%
| 555
| 95%
|
| 已实施待批(CBE)的生产补充申请
| 1147
| 93%
| 1099
| 97%
|
CDER NDA/BLA 立卷与批准数
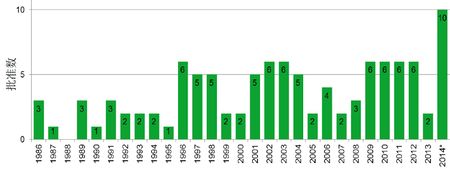
1986-2014年生物制品获批情况
1983-2014年罕见病用药获批情况
2000-2014年新分子实体药品获批情况
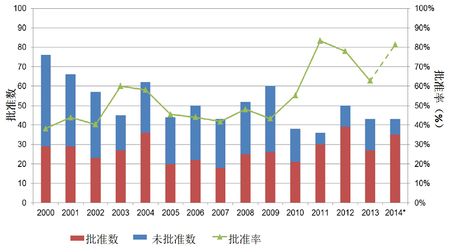
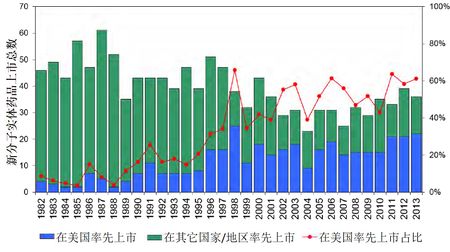
1982-2014新分子实体药品在美国率先上市占比
PDUFA I-PDUFA V期间CDER新分子实体药品中位批准时间
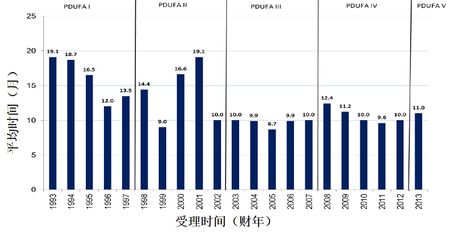
(数据来源:FDA CDER新药办公室主任 John K. Jenkins博士2014年FDA/CMS峰会报告)
识林TMwww.shilinx.com版权所有,未经许可不得转载。如需使用请联系admin@shilinx.com
岗位必读建议: - 注册:了解FDASIA对药物和医疗设备注册审批流程的影响。
- 研发:关注创新药和仿制药的用户费用要求,以及对儿科药物开发的支持。
- QA:确保产品质量和安全性符合FDASIA规定的标准。
文件适用范围:
本文适用于美国境内的创新药、医疗设备、仿制药和生物类似药的注册分类,由美国食品药品监督管理局(FDA)发布,适用于Biotech、大型药企、跨国药企等各类企业。 文件要点总结: - 用户费用授权:FDASIA授权FDA从行业收取用户费用,以资助创新药、医疗器械、仿制药和生物类似药的审查工作。
- 儿科药物开发鼓励:该法案重新授权两个鼓励儿科药物开发的项目。
- PDUFA和MDUFA的第五次和第三次授权:这是处方药用户费用法案(PDUFA)的第五次授权和医疗器械用户费用法案(MDUFA)的第三次授权。
- 审查流程的稳定性和可靠性:通过这些用户费用计划,确保了审查人员队伍的稳定和审查流程的可靠性。
- 仿制药和生物类似药的用户费用计划:新计划建立在PDUFA和MDUFA成功的基础上,为仿制药和生物类似药的审查提供资金。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |






