|
首页
>
资讯
>
国内药政每周导读:2022.03.14-03.20
出自识林
国内药政每周导读:2022.03.14-03.20
2022-03-21
【监管综合】
3.18,NMPA发布《药品监督管理统计报告2021年第三季度》
本报告期为2021年1月1日至9月30日,包含药品受理、注册审批、国产和进口药品批准文号、生产日常监管、地方抽检等大量数据。
3.18,中检院发布《国家药品抽检年报2021》
抽检一向是业界颇为关注的上市后监管行动,尤其是无菌制剂和中药企业,且看2021年总体数据如何:
— 2021年国家药品抽检共抽取制剂产品与中药饮片品种139个,包括化学药品81个、中成药46个、中药饮片9个、生物制品3个,其中国家基本药物品种53个;共抽检样品17856批次,包括生产环节3965批次、经营环节12409批次、使用环节1250批次和口岸环节232批次,涉及1113家药品生产企业、2085家药品经营企业和486家药品使用单位。
— 2021年国家药品抽检共抽检制剂产品15899批次。经检验,15861批次产品符合规定,38批次产品不符合规定。抽检的130个品种中,全部样品符合规定的制剂产品有108个,共12419批次。 其中,化学药品有70个品种7674批次,中成药有35个品种4681批次,生物制品有3个品种64批次。
— 化学药品不符合规定项目包括性状、鉴别、检查和含量测定,不符合规定产品数量依次为1、4、14和2批次,分别占全部不符合规定项目的4.8%、19.0%、66.7%和9.5%。不符合规定项目中检查项占比最大,涉及“有关物质”、“溶出度”等检验项目,不符合规定原因主要与生产工艺中压片强度、原辅料混合程度、运输储存过程中的温度影响等相关因素有关。
— 中成药不符合规定项目主要涉及鉴别、检查、含量测定等,不符合规定样品批次依次为9批次、7批次和4批次,分别占全部不符合规定项目的45.0%、35.0%和20.0%。
— 生物制品3个品种64批次所检项目均符合规定,合格率为100%。
— 进口药品抽检590批次,涉及4个剂型,其中口岸环节232批次、生产环节14批次、经营环节288批次、使用环节56批次。经检验,所检项目均符合规定,合格率为100%。
— 中药饮片不符合规定项目主要涉及总灰分(2批次)、性状(18批次)、杂质(11批次)、鉴别(1批次)和含量测定(3批次)等方面,分别占全部不符合规定项目的5.7%、51.4%、31.4%、2.9%和8.6%。
【临床研究】
3.14,CDE发布《化学药品改良型新药临床试验技术指导原则问与答》第1版(征求意见稿)
自2020年12月NMPA发布《化学药品改良型新药临床试验技术指导原则》以来,CDE在沟通交流和各类咨询通道中收到许多来自研发企业和科研机构的问题,这些问题主要集中在对指导原则中具体技术标准和审评原则的理解方面。
为进一步促进改良型新药研发,CDE特发此文,罗列8个共性问题。
这些共性问题涉及“提高有效性”,“开发新适应症”,“改善安全性”以及“提高依从性”。
企业提得相当具体,如某企业:“发现其中一个已获批上市的手性分子,它的一个异构体虽然有效,但存在明确的安全性问题,去除该异构体,提高了安全性,这个改良方式算不算改良型新药?需要开展哪些临床试验?”
CDE的答复也足够明确:“如果确认拟去除的异构体存在明确的安全性问题,认可这样的改良方式”,并且还举例说明:“抗过敏药物西替利嗪,右西替利嗪可与大脑中的受体结合,从而导致镇静和嗜睡的不良反应,去除右西替利嗪,保留左西替利嗪发挥抗过敏作用。”
如此问答,能够节省企业大量时间,也易于控制成本和风险,多多益善。
识林用户可查阅原文和配套法规,结合自身项目,深入理解。
此外,该问答征求意见截至4月14日,如有疑问或意见,还可进一步澄清。
【CMC】
3.14,药典委发布《中国药典》2020年版微生物相关通用技术要求共性问题的答复(一)
该文包含7个问题,涉及药典四部通则中的1101无菌检查法和9203药品微生物实验室质量管理指导原则。药典委的回复是对指导原则的进一步完善。
【变更】
3.14,陕西省局发布《药品上市后变更管理常见问题解答(二)》
本文共计9个问题:
一、省局备案类变更,在国家局网站查询到已公示是否可以立即实施?
二、药品说明书、标签变更属于重大变更事项的持有人如何申请?
三、属于药品说明书、标签中等变更事项有哪些情形?
四、申请批准或备案类的药品上市后变更,如涉及药品说明书或标签修订的,如何申请?
五、报告类的药品上市后变更,如涉及药品说明书或标签修订的,持有人如何处理?
六、某药品申报了药品上市许可持有人变更并已获批,现准备对该品种的说明书和标签中上市许可持有人信息进行修订,是否需要备案?
七、某产品变更有效期进行备案后,是否还需要进行修订说明书、包装标签的备案?
八、企业申请生产场地变更,是否需要进行修订说明书、包装标签的备案?已经完成备案,未同时写明变更后说明书、标签的,如何处理?
九、药品在同一个生产地址内由一个生产车间变更至另一车间,是否需要申报注册事项变更?
变更情形复杂,所以,即使法规体系已经相当完善,企业仍需要这类具体的问答指导。
目前识林已在“省局药品上市后变更管理实施细则”页面中收录28个省市自治区药监局的上市后变更相关法规,其中有海南、陕西、江苏、山东发布问答类文件,可供参考。
此外,识林用户可阅读主题词“变更”,系统了解法规要求,还可通过“中国化学药品上市后变更思维导图速查工具”,方便快捷检索变更对应的研究要求。
【医疗器械】
3.15,CDR发布《国家医疗器械不良事件监测年度报告2021年》
CDR(不良反应监测中心)的官方年度总结报告,医疗器械企业可关注。
部分重点数据:
— 2021年,国家医疗器械不良事件监测信息系统接收到医疗器械不良事件报告65万余份,比上年增加21.39%;每百万人口平均医疗器械不良事件报告数为461份,比上年增加14.68%;
— CDR收到的报告中,使用单位上报562,928份,占报告总数的86.52%;注册人上报14,853份,占报告总数的2.28%;经营企业上报72,567份,占报告总数的11.15%;其他来源的报告347份,占报告总数的0.05%。
— CDR收到的报告中,伤害程度为死亡的报告163份,占报告总数的0.03%;伤害程度为严重伤害的报告36,610份,占报告总数的5.63%;伤害程度为其他的报告613,922份,占报告总数的94.35%。
另外,《国家药品不良反应监测年度报告》预计也将在近日发布。
3.18,NMPA已批准17个新冠病毒抗原检测试剂
结合最新发布的《2022年03月18日医疗器械批准证明文件(准产)待领取信息》,国家药监局已批准17个新冠病毒抗原检测试剂产品。
【中药】
NMPA及相关部门联合发布新GAP《中药材生产质量管理规范》
暌违20年,GAP(Good Agricultural Practice)终于迎来现行版。
这20年中,2002年4月《中药材生产质量管理规范(试行)》发布,2016年取消GAP认证,2017年10月发修订稿,2018年发征求意见稿,到了2021年4月,SAMR在新版GAP尚未落地时,就直接废止了2002年版(见“SAMR 关于废止和修改部分规章的决定”)。
中药材是中药的原料。对于化药,法规指南众多,API工厂通常有完善的质量管理体系,且合规性有药监部门监管,制剂厂商主要是通过审计和质量协议来管控。由于中药的特殊性,中药制剂企业与中药材的联系尤为紧密,因此GAP是整个中药行业都应关注的。
相比2002版,2022现行版内容增加了87条,变动巨大,业界需要从根本上改变思想观念和日常操作。
将现行版与2018年的征求意见稿进行对比更具意义,4年之间,到底有哪些增删,体现了哪些监管导向和风险考量,值得深思。
识林兹提供一些显著的变化,供参考:
— 新增“野生中药材的采收加工可参考本规范”,让天然环境下获得的药材有法可依。此外还增加了一些“野生抚育”和“野生栽培”相关的条款;
— 新增“根据生产实际情况确定质量控制指标”,可包括“......指纹或者特征图谱...药材农药残留或者兽药残留、重金属及有害元素...等”。之前的提法是“原则上应当有”。这是否意味着药材标准更加合理,可以避免一些成本很高的过度检测?
— 新增“检验可以自行检验,也可以委托第三方或中药材使用单位检验。”
— “批”的定义中,新增要求“种子种苗或其它繁殖材料来源相同”。
以上仅为导读,企业可至官网或识林查阅原文,并用“页面比对”工具阅读花脸稿。
【新药批准和报产】
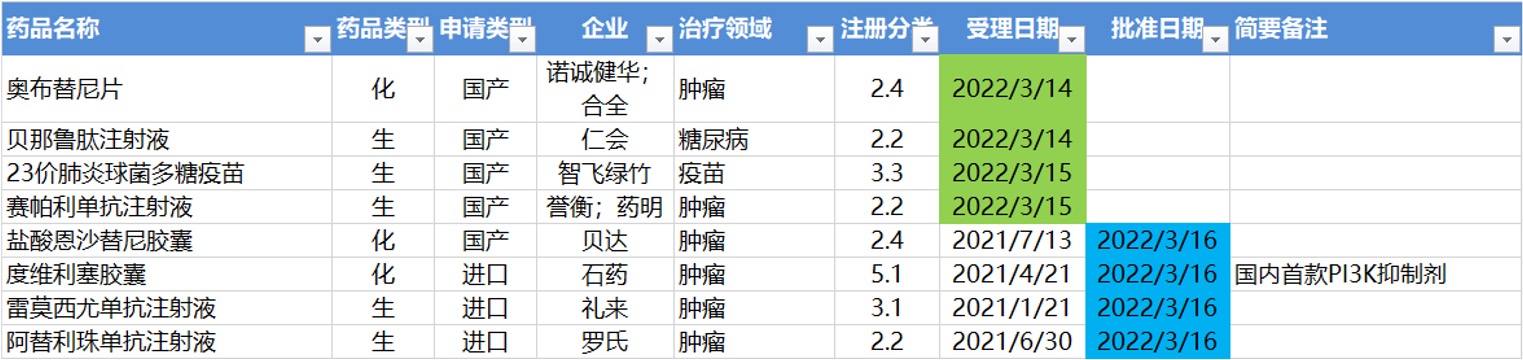
3.14-3.20,NMPA发布4个新药批准,包括国内首个PI3K抑制剂,CDE受理4个新药上市
NMPA发布4个新药批准信息,包括石药引进的国内首个PI3K抑制剂度维利塞胶囊。
CDE受理4个新药上市申请。
注:仅列出新药(包括改良型新药和生物类似药)的上市申请和批准上市信息。在“以临床价值为导向”的背景下,申请上市以及获批的品种,其适应症、临床研究策略、注册路径,都值得业界关注和分析。
作者:识林-实木
识林®版权所有,未经许可不得转载。如需使用请联系 admin@shilinx.com 。
岗位必读建议: - 研发(R&D):应深入理解改良型新药的临床优势和试验设计原则,以指导新药研发方向。
- 临床(Clinical):必须熟悉临床试验设计与评价的指导原则,确保试验方案的科学性和合规性。
- 注册(Regulatory Affairs, RA):需掌握改良型新药的注册要求和申报资料要求,以促进顺利注册。
- 质量管理(QA):应监督临床试验的质量管理,确保试验符合GCP标准。
文件适用范围:
本文适用于化学药品改良型新药的临床试验设计和评价,包括境内外已上市但境内未上市的改良型化学药品。主要针对创新药或仿制药,不包括复方制剂。发布机构为中国药品审评中心(CDE)。 文件要点总结: - 临床优势定义:明确了改良型新药应具备的临床优势,包括提高疗效、改善安全性和提高依从性。
- 有效性优势路径:提出了与已上市药品相同或不同的目标适应症时,如何通过临床试验证明有效性优势。
- 安全性优势考量:强调了在不降低疗效的前提下,如何通过改良API结构或剂型等途径显著改善安全性。
- 依从性优势策略:讨论了通过改变给药途径或剂型提高患者用药依从性的方法。
- 其他优势及沟通:鼓励与药品审评中心沟通,针对未涵盖的改良型新药优势和试验设计进行讨论。
以上仅为部分要点,请阅读原文,深入理解监管要求。 【文件概要】
本文整合了《中药材生产质量管理规范》(GAP)的全面要求,涵盖中药材生产全周期的质量管理体系。规范明确了中药材生产企业的基本要求,包括种植(含生态种植、野生抚育和仿野生栽培)、养殖、采收、产地加工、包装、储运等环节的技术规程和管理标准。文件强调质量追溯体系的建立,要求企业实现从种子种苗到成品发运的全过程可追溯,并鼓励运用现代信息技术。规范对生产基地选址、种子种苗管理、农业投入品使用、病虫害防治、采收加工方法、包装标签、贮存条件等提出具体技术要求,禁止使用高毒农药、生长调节剂及硫磺熏蒸等违规操作。文件还要求企业制定内审制度、投诉召回机制,并明确了中药材批的定义、放行程序和文件管理要求。 【适用范围】
本文适用于中国境内从事中药材生产的各类企业,包括种植/养殖专业合作社、中药饮片生产企业及中成药上市许可持有人。规范针对药用植物、药用动物来源的中药材,涵盖种植、养殖、野生抚育和仿野生栽培等生产方式,适用于中药饮片和制剂生产的药用原料。 【影响评估】
本文强制要求中药材生产企业建立全流程质量管理体系,可能增加生产成本,但有助于提升药材质量合规性。中药饮片和中成药企业需优先采购符合GAP的药材,并可能需调整供应商审核流程。规范鼓励产地自建基地,可能推动行业纵向整合。 【实施建议】 - 必读岗位:
- 注册:更新供应商审核标准,确保药材来源符合GAP要求;标签中“药材符合GAP要求”的标示需合规。
- QA/QC:建立药材质量追溯体系,强化批次检验与留样管理;监督农业投入品使用合规性。
- 生产(种植/养殖):制定技术规程,重点管控种子种苗、采收加工、贮存环节;禁止使用违禁农药/生长调节剂。
- 采购:优先选择GAP认证供应商,完善合同质量条款。
以上仅为部分要点,请阅读原文,深入理解监管要求。 适用岗位: - QC:必读,负责实验室日常质量管理和微生物检测工作。
- QA:必读,负责监督和审核微生物实验室的质量管理流程。
- 研发:必读,涉及微生物质量控制的研发项目需遵循此指导原则。
工作建议: - QC:确保实验室操作符合微生物检测的质量管理要求,定期进行自检和质量控制。
- QA:审核微生物实验室的质量管理体系,确保符合指导原则要求。
- 研发:在研发过程中,特别关注微生物质量控制的合规性,确保研发数据的准确性和可靠性。
适用范围:
本文适用于化学药、生物制品、疫苗和中药等药品类型,涉及创新药、仿制药、生物类似药、原料药等注册分类,适用于中国境内的Biotech、大型药企、跨国药企、CRO和CDMO等企业类别。 文件要点总结: - 质量管理体系建设:明确要求建立和维护微生物实验室的全面质量管理体系。
- 人员资质与培训:强调微生物实验室人员必须具备相应的资质,并接受定期培训。
- 实验室环境控制:规定实验室环境应符合微生物检测的要求,包括温湿度控制和洁净度等级。
- 设备与材料管理:强调对实验室使用的设备和材料进行严格的管理和定期校准。
- 检测方法验证:要求对微生物检测方法进行验证,确保检测结果的准确性和可靠性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 法规指南解读:中国药典2020_四部_1101_无菌检查法 适用岗位: - QA(质量保证):必读,需确保无菌检查流程符合药典要求。
- QC(质量控制)实验室人员:必读,负责执行无菌检查操作。
- 生产部门:了解,需知晓无菌操作的重要性。
工作建议: - QA应定期审核无菌检查流程,确保符合最新法规要求。
- QC实验室人员应严格遵循无菌操作规程,保证检验结果的准确性。
- 生产部门应加强无菌操作培训,减少生产过程中的微生物污染风险。
文件适用范围:
本文适用于化学药品、生物制品、医疗器械等的无菌检查,包括原料药、辅料等。适用于在中国注册的各类药企,包括Biotech、大型药企、跨国药企等。 要点总结: - 无菌检查环境要求:强调无菌检查应在无菌条件下进行,环境需定期进行洁净度确认。
- 培养基制备与灭菌:规定了硫乙醇酸盐流体培养基和胰酪大豆胨液体培养基的制备方法和灭菌程序。
- 培养基适用性检查:要求对培养基进行无菌性和灵敏度检查,以确保培养基符合要求。
- 方法适用性试验:进行无菌检查前,必须进行方法适用性试验,以确认所采用的无菌检查方法适合于该产品。
- 供试品的无菌检查:详细描述了薄膜过滤法和直接接种法的操作步骤,以及阳性和阴性对照的要求。
以上仅为部分要点,请阅读原文,深入理解监管要求。 必读岗位及工作建议: - QA:负责确保质量管理体系的实施和监督,建议定期审查和更新质量管理体系文件。
- 生产:确保生产过程符合质量管理体系要求,建议参与设备和工艺管理的持续改进。
- 研发:在产品设计和开发阶段考虑质量管理体系要求,建议与QA紧密合作以确保合规性。
适用范围:
本文适用于涉及化学药、生物制品、疫苗和中药等药品类型的企业,包括创新药、仿制药、生物类似药和原料药等注册分类。适用于不同规模的企业,如Biotech、大型药企、跨国药企、CRO和CDMO等,由相关药品监管机构发布。 文件要点总结: - 质量管理体系概述:明确了质量管理体系的发展、基本概念及其相互关系,强调了高层管理者在质量方针、目标和计划制定中的关键作用。
- 产品质量实现要素:涵盖了机构与人员、厂房设施、设备、物料与产品、工艺管理等关键要素,特别指出了人员培训和设备生命周期管理的重要性。
- 质量保证要素:包括变更管理、偏差管理、产品质量回顾、投诉和召回管理,强调了CAPA系统在持续改进中的作用。
- 质量风险管理:介绍了质量风险管理的职责、模式图、流程和步骤,以及在企业和管理机构中的应用。
- 质量管理系统文件:规定了文件体系结构、生命周期和种类,强调了文件管理在确保质量管理体系有效运行中的重要性。
以上仅为部分要点,请阅读原文,深入理解监管要求。 |